
Semaglutide
Glucagon-like Peptide-1 (GLP-1) Receptor Agonist
Ozempic, Wegovy, Rybelsus (oral)
Contents
Sequence:
H-His-Aib-Glu-Gly-Thr-Phe-Thr-Ser-Asp-Val-Ser-Ser-Tyr-Leu-Glu-Gly-Gln-Ala-Ala-Lys-Glu-Phe-Ile-Ala-Trp-Leu-Val-Arg-Gly-Arg-Gly-OH
Molar Weight:
4113.6 g/mol
Molecular Formula:
C187H291N45O59
Most Frequent Uses:
- Semaglutide is a glucagon-like peptide 1 (GLP-1) receptor agonist indicated as an adjunct to diet and exercise to improve glycemic control in adults with type 2 diabetes mellitus.
Dosage:
Oral
- Initially 3 mg once a day for 30 days
- Then increase to 7mg once daily for 30 days
- Then 14 mg once a day target dosage if warranted
- Swallow whole on an empty stomach, at least 30min before eating
- No dosage adjustment in hepatic or renal impairment
- Oral semaglutidecan be initiated 7 days after the last dose of injectable semaglutide administered
- There is no equivalent dose of oral semaglutidefor subcutaneous semaglutide 1 mg weekly per manufacturer
SubQ
For T2D or pre-diabetes – SubQ
- Start at 0.25 mg (250mcg) once weekly.
- After 4 weeks, increase the dose to 0.5 mg (500mcg) once weekly.
- If after at least 4 weeks additional glycemic control is needed, increase to 1 mg once weekly
- Administer once weekly at any time of day, with or without meals; semaglutide should not be administered daily.
- If a dose is missed administer within 5 days of missed dose
- Inject subcutaneously in the abdomen, thigh, or upper arm; rotate weekly
- Semaglutide injections should appear clear and colorless – Do not use semaglutide if particulate matter and coloration is seen.
For Weight Loss – SubQ
- Start with 100mcg weekly
- If no nausea, increase 50mcg-100mcg weekly until 250-500mcg obtained or until clinical endpoints achieved (no > than 500mcg weekly).
- Final dosage is individual and depends on degree of weight loss needed and clinical presentation.
- Some individuals may require T2D and pre-diabetes dosages.
General Dosage Comments
- Mono- or combo-therapy when metformin is not tolerated
- When semaglutide is added to existing metformin therapy, the current dose of metformin can be continued unchanged.
- An increased risk of hypoglycemia was seen with concomitant use of SU or basal insulin with semaglutide. When semaglutide is added to existing therapy of a sulfonylurea or insulin, a reduction in the dose of sulfonylurea or insulin should be considered to reduce the risk of hypoglycemia. In clinical trials, insulin dose was decreased by 20% at onset of semaglutide treatment.
- * Can add oral Vitamin B12 (methylcobalamin for best absorption), 1000mcg (1mg) concurrent with semaglutide dose) – reduces nausea and GI side effects
Safety and Potential Side Effects/Contraindications:
- Semaglutide is reported safe in recommended dosages.
WARNING: RISK OF THYROID C-CELL TUMORS
|
- Semaglutide is contraindicated in patients with a personal or family history of medullary thyroid carcinoma (MTC) or in patients with Multiple Endocrine Neoplasia syndrome type 2 (MEN 2).
- Contraindicated in patients with hypersensitivity to Semaglutide.
- The most common adverse reactions, reported in ≥5% of patients treated with semaglutide are:
- Nausea/vomiting
- Diarrhea
- Abdominal pain
- Constipation
- Semaglutide should not be used during pregnancy or breastfeeding.
Further Contraindications:
- Pancreatitis: Has been reported in clinical trials. Discontinue promptly if pancreatitis is suspected. Do not restart if pancreatitis is confirmed
- Diabetic Retinopathy Complications: Has been reported in a clinical trial. Patients with a history of diabetic retinopathy should be monitored
- Hypoglycemia: When semaglutide is used with an insulin secretagogue or insulin, consider lowering the dose of the secretagogue or insulin to reduce the risk of hypoglycemia
- Acute Kidney Injury: Monitor renal function in patients with renal impairment reporting severe adverse gastrointestinal reactions
- Hypersensitivity Reactions: Discontinue semaglutide if suspected and promptly seek medical advice
- Macrovascular outcomes: There have been no clinical studies establishing conclusive evidence of macrovascular risk reduction with semaglutide.
Description
(*from OZEMPIC product monograph, https://www.novonordisk.ca/content/dam/nncorp/ca/en/products/ozempic-product-monograph.pdf)
Mechanism of Action
Semaglutide is a GLP-1 analogue with 94% sequence homology to human GLP-1. Semaglutide acts as a GLP-1 receptor agonist that selectively binds to and activates the GLP-1 receptor, the target for native GLP-1. GLP-1 is a physiological hormone that has multiple actions on glucose, mediated by the GLP-1 receptors. The principal mechanism of protraction resulting in the long half-life of semaglutide is albumin binding, which results in decreased renal clearance and protection from metabolic degradation. Furthermore, semaglutide is stabilized against degradation by the DPP-4 enzyme. Semaglutide reduces blood glucose through a mechanism where it stimulates insulin secretion and lowers glucagon secretion, both in a glucose-dependent manner. Thus, when blood glucose is high, insulin secretion is stimulated, and glucagon secretion is inhibited. The mechanism of blood glucose lowering also involves a minor delay in gastric emptying in the early postprandial phase.
Pharmacodynamics
Semaglutide lowers fasting and postprandial blood glucose and reduces body weight. All pharmacodynamic evaluations were performed after 12 weeks of treatment (including dose escalation) at steady state with semaglutide 1 mg.
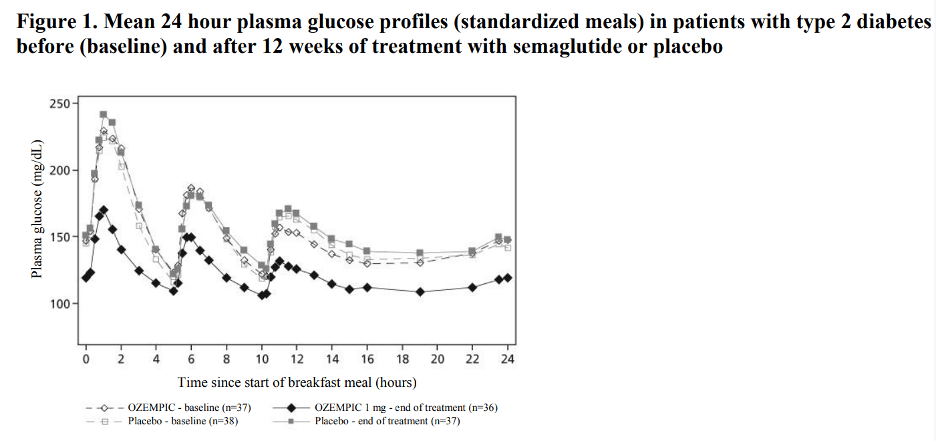
Fasting and Postprandial Glucose
Semaglutide reduces fasting and postprandial glucose concentrations. In patients with type 2 diabetes, treatment with semaglutide 1 mg resulted in reductions in glucose in terms of absolute change from baseline and relative reduction compared to placebo of 29 mg/dL (22%) for fasting glucose, 74 mg/dL (36%) for 2 hour postprandial glucose, and 30 mg/dL (22%) for mean 24 hour glucose concentration (see Figure 1).
Insulin Secretion
Both first-and second-phase insulin secretion are increased in patients with type 2 diabetes treated with semaglutide compared with placebo.
Glucagon Secretion
Semaglutide lowers the fasting and postprandial glucagon concentrations. In patients with type 2 diabetes, treatment with semaglutide resulted in the following relative reductions in glucagon compared to placebo, fasting glucagon (8%), postprandial glucagon response (14-15%), and mean 24 hour glucagon concentration (12%).
Glucose dependent insulin and glucagon secretion
Semaglutide lowers high blood glucose concentrations by stimulating insulin secretion and lowering glucagon secretion in a glucose-dependent manner. With semaglutide, the insulin secretion rate in patients with type 2 diabetes was similar to that of healthy subjects (see Figure 2).
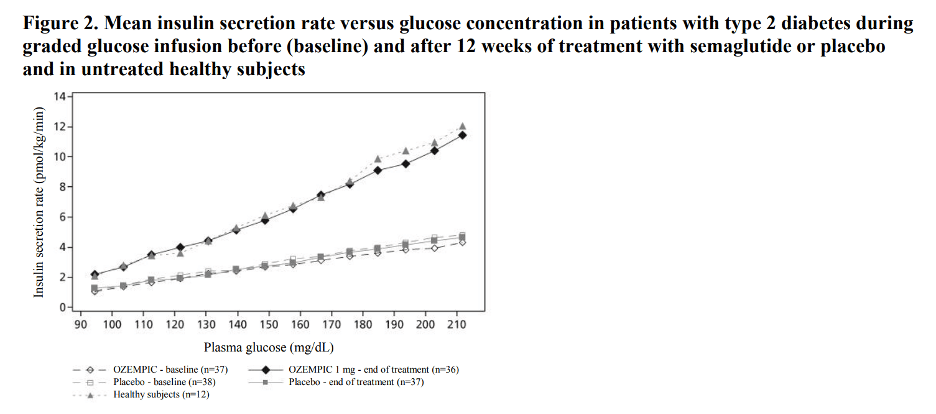
During induced hypoglycemia, semaglutide did not alter the counter regulatory responses of increased glucagon compared to placebo and did not impair the decrease of C-peptide in patients with type 2 diabetes.
Gastric emptying
Semaglutide causes a delay of early postprandial gastric emptying, thereby reducing the rate at which glucose appears in the circulation postprandially.
Cardiac electrophysiology (QTc)
The effect of semaglutide on cardiac repolarization was tested in a thorough QTc trial. At a dose 1.5 times the maximum recommended dose, semaglutide does not prolong QTc intervals to any clinically relevant extent.
Pharmacokinetics
Absorption
Absolute bioavailability of semaglutide is 89%. Maximum concentration of semaglutide is reached 1 to 3 days post dose. Similar exposure is achieved with subcutaneous administration of semaglutide in the abdomen, thigh, or upper arm. In patients with type 2 diabetes, semaglutide exposure increases in a dose-proportional manner for once-weekly doses of 0.5 mg and 1 mg. Steady-state exposure is achieved following 4-5 weeks of once-weekly administration. In patients with type 2 diabetes, the mean population-PK estimated steady-state concentrations following once weekly subcutaneous administration of 0.5 mg and 1 mg semaglutide were approximately 65.0 ng/mL and 123.0 ng/mL, respectively.
Distribution
The mean apparent volume of distribution of semaglutide following subcutaneous administration in patients with type 2 diabetes is approximately 12.5 L. Semaglutide is extensively bound to plasma albumin (>99%).
Elimination
The apparent clearance of semaglutide in patients with type 2 diabetes is approximately 0.05 L/h. With an elimination half-life of approximately 1 week, semaglutide will be present in the circulation for about 5 weeks after the last dose.
- Metabolism
The primary route of elimination for semaglutide is metabolism following proteolytic cleavage of the peptide backbone and sequential beta-oxidation of the fatty acid sidechain.
- Excretion
The primary excretion routes of semaglutide-related material is via the urine and feces. Approximately 3% of the dose is excreted in the urine as intact semaglutide.
Specific Populations
Based on a population pharmacokinetic analysis, age, sex, race, and ethnicity, and renal impairment do not have a clinically meaningful effect on the pharmacokinetics of semaglutide. The exposure of semaglutide decreases with an increase in body weight. However, semaglutide doses of 0.5 mg and 1 mg provide adequate systemic exposure over the body weight range of 40-198 kg evaluated in the clinical trials. The effects of intrinsic factors on the pharmacokinetics of semaglutide are shown in Figure 3.

Patients with Renal impairment
Renal impairment does not impact the pharmacokinetics of semaglutide in a clinically relevant manner. This was shown in a study with a single dose of 0.5 mg semaglutide in patients with different degrees of renal impairment (mild, moderate, severe, ESRD) compared with subjects with normal renal function. This was also shown for subjects with both type 2 diabetes and renal impairment based on data from clinical studies (Figure 3).
Patients with Hepatic impairment
Hepatic impairment does not have any impact on the exposure of semaglutide. The pharmacokinetics of semaglutide were evaluated in patients with different degrees of hepatic impairment (mild, moderate, severe) compared with subjects with normal hepatic function in a study with a single-dose of 0.5 mg semaglutide.
Pediatric Patients
Semaglutide has not been studied in pediatric patients.
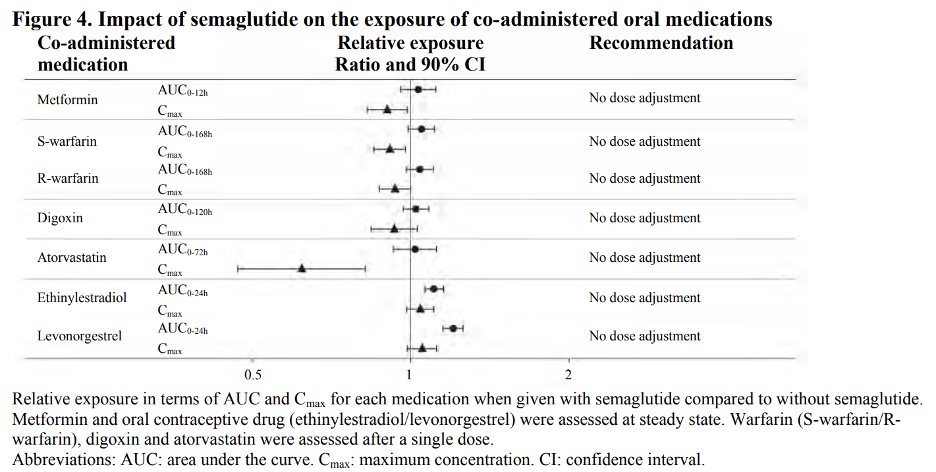
Drug Interaction Studies
In vitro studies have shown very low potential for semaglutide to inhibit or induce CYP enzymes, and to inhibit drug transporters. The delay of gastric emptying with semaglutide may influence the absorption of concomitantly administered oral medicinal products. The potential effect of semaglutide on the absorption of co-administered oral medications was studied in trials at semaglutide 1 mg steady-state exposure. No clinically relevant drug-drug interaction with semaglutide (Figure 4) was observed based on the evaluated medications; therefore, no dose adjustment is required when co-administered with semaglutide.
Clinical Study Summary
Semaglutide has been studied as monotherapy and in combination with metformin, metformin and sulfonylureas, metformin and/or thiazolidinedione, and basal insulin in patients with type 2 diabetes mellitus. The efficacy of Semaglutide was compared with placebo, sitagliptin, exenatide extended-release (ER), and insulin glargine. Most trials evaluated the use of Semaglutide 0.5 mg, and 1 mg, with the exception of the trial comparing Semaglutide and exenatide ER where only the 1 mg dose was studied. In patients with type 2 diabetes mellitus, Semaglutide produced clinically relevant reduction from baseline in HbA1c compared with placebo. The efficacy of Semaglutide was not impacted by age, gender, race, ethnicity, BMI at baseline, body weight (kg) at baseline, diabetes duration and level of renal function impairment.
Monotherapy Use of Semaglutide in Patients with Type 2 Diabetes Mellitus
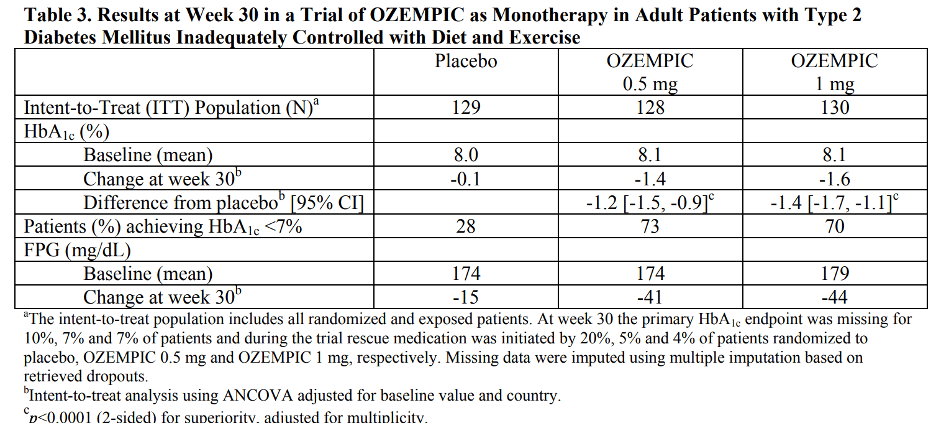
In a 30-week double-blind trial (NCT02054897), 388 patients with type 2 diabetes mellitus inadequately controlled with diet and exercise were randomized to Semaglutide 0.5 mg or Semaglutide 1 mg once weekly or placebo. Patients had a mean age of 54 years and 54% were men. The mean duration of type 2 diabetes was 4.2 years, and the mean BMI was 33 kg/m2 . Overall, 64% were White, 8% were Black or African American, and 21% were Asian; 30% identified as Hispanic or Latino ethnicity. Monotherapy with Semaglutide 0.5 mg and 1 mg once weekly for 30 weeks resulted in a statistically significant reduction in HbA1c compared with placebo (see Table 3).
The mean baseline body weight was 89.1 kg, 89.8 kg, 96.9 kg in the placebo, semaglutide 0.5 mg, and semaglutide 1 mg arms, respectively. The mean changes from baseline to week 30 were -1.2 kg, -3.8 kg and -4.7 kg in the placebo, semaglutide 0.5 mg, and semaglutide 1 mg arms, respectively. The difference from placebo (95% CI) for semaglutide 0.5 mg was -2.6 kg (-3.8, -1.5), and for semaglutide 1 mg was -3.5 kg (-4.8, -2.2).
Clinical Research
IPS Level of Evidence
IPS Clinical Pharmacists have developed a method of ranking the studies so that the practitioner can easily discern the level of evidence this study provides to the topic. Levels 1-8 are listed below:
| Level of Evidence | Description | |
| X | Level 1 | FDA Approved Drug studies |
| X | Level 2 | Evidence obtained from systematic review and/or meta-analyses of studies including RCTs and other human studies |
| X | Level 3 | Evidence obtained from a RCT |
| Level 4 | Evidence obtained from a study without randomization | |
| Level 5 | Evidence obtained from case reports | |
| Level 6 | Evidence obtained from in vitro human studies | |
| X | Level 7 | Evidence obtained from laboratory animal studies |
| X | Level 8 | Evidence obtained from Opinions or Reviews |
Levels
Level 1
Wilding JPH, et al. Once-Weekly Semaglutide in Adults with Overweight or Obesity. N EJM. 2021;384:989-1002.
Abstract
Background: Obesity is a global health challenge with few pharmacologic options. Whether adults with obesity can achieve weight loss with once-weekly semaglutide at a dose of 2.4 mg as an adjunct to lifestyle intervention has not been confirmed.
Methods: In this double-blind trial, we enrolled 1961 adults with a body-mass index (the weight in kilograms divided by the square of the height in meters) of 30 or greater (≥27 in persons with ≥1 weight-related coexisting condition), who did not have diabetes, and randomly assigned them, in a 2:1 ratio, to 68 weeks of treatment with once-weekly subcutaneous semaglutide (at a dose of 2.4 mg) or placebo, plus lifestyle intervention. The coprimary end points were the percentage change in body weight and weight reduction of at least 5%. The primary estimand (a precise description of the treatment effect reflecting the objective of the clinical trial) assessed effects regardless of treatment discontinuation or rescue interventions.
Results: The mean change in body weight from baseline to week 68 was -14.9% in the semaglutide group as compared with -2.4% with placebo, for an estimated treatment difference of -12.4 percentage points (95% confidence interval [CI], -13.4 to -11.5; P<0.001). More participants in the semaglutide group than in the placebo group achieved weight reductions of 5% or more (1047 participants [86.4%] vs. 182 [31.5%]), 10% or more (838 [69.1%] vs. 69 [12.0%]), and 15% or more (612 [50.5%] vs. 28 [4.9%]) at week 68 (P<0.001 for all three comparisons of odds). The change in body weight from baseline to week 68 was -15.3 kg in the semaglutide group as compared with -2.6 kg in the placebo group (estimated treatment difference, -12.7 kg; 95% CI, -13.7 to -11.7). Participants who received semaglutide had a greater improvement with respect to cardiometabolic risk factors and a greater increase in participant-reported physical functioning from baseline than those who received placebo. Nausea and diarrhea were the most common adverse events with semaglutide; they were typically transient and mild-to-moderate in severity and subsided with time. More participants in the semaglutide group than in the placebo group discontinued treatment owing to gastrointestinal events (59 [4.5%] vs. 5 [0.8%]).
Conclusions: In participants with overweight or obesity, 2.4 mg of semaglutide once weekly plus lifestyle intervention was associated with sustained, clinically relevant reduction in body weight. (Funded by Novo Nordisk; STEP 1 ClinicalTrials.gov number, NCT03548935).
https://www.nejm.org/doi/full/10.1056/NEJMoa2032183
Aroda VR, et al. Comparative efficacy, safety, and cardiovascular outcomes with once-weekly subcutaneous semaglutide in the treatment of type 2 diabetes: Insights from the SUSTAIN 1-7 trials. Diabetes Metab. 2019;45(5):409-18.
Abstract
In individuals with type 2 diabetes, glycaemic control and cardiovascular risk factor management reduces the likelihood of late-stage diabetic complications. Guidelines recommend treatment goals targeting HbA1c, body weight, blood pressure, and low-density lipoprotein cholesterol. Development of new treatments for type 2 diabetes requires an understanding of their mechanism and efficacy, as well as their relative effects compared to other treatment choices, plus demonstration of cardiovascular safety. Subcutaneous semaglutide is a glucagon-like peptide-1 receptor agonist currently approved in several countries for once-weekly treatment of type 2 diabetes. Semaglutide works via the incretin pathway, stimulating insulin and inhibiting glucagon secretion from the pancreatic islets, leading to lower blood glucose levels. Semaglutide also decreases energy intake by reducing appetite and food cravings, and lowering relative preference for fatty, energy-dense foods. Semaglutide was evaluated in the SUSTAIN clinical trial programme in over 8000 patients across the spectrum of type 2 diabetes. This review details the efficacy and safety profile of semaglutide in the SUSTAIN 1-5 and 7 trials, and its cardiovascular safety profile in the SUSTAIN 6 trial. Semaglutide consistently demonstrated superior and sustained glycemic control and weight loss vs. all comparators evaluated. In SUSTAIN 6, involving patients at high risk of cardiovascular disease, semaglutide significantly decreased the occurrence of cardiovascular events compared with placebo/standard of care (hazard ratio 0.74, P < 0.001 for non-inferiority). Through a comprehensive phase 3 clinical trial program, we have a detailed understanding of semaglutide’s efficacy, safety, cardiovascular effects and comparative role in the treatment of type 2 diabetes.
https://www.gwern.net/docs/longevity/semaglutide/2019-aroda.pdf
O’Neil PM, et al. Efficacy and safety of semaglutide compared with liraglutide and placebo for weight loss in patients with obesity: a randomised, double-blind, placebo and active controlled, dose-ranging, phase 2 trial. Lancet. 2018;92(10148):637-49.
Abstract
Background: Obesity is a major public health issue, and new pharmaceuticals for weight management are needed. Therefore, we evaluated the efficacy and safety of the glucagon-like peptide-1 (GLP-1) analogue semaglutide in comparison with liraglutide and a placebo in promoting weight loss.
Methods: We did a randomised, double-blind, placebo and active controlled, multicentre, dose-ranging, phase 2 trial. The study was done in eight countries involving 71 clinical sites. Eligible participants were adults (≥18 years) without diabetes and with a body-mass index (BMI) of 30 kg/m2 or more. We randomly assigned participants (6:1) to each active treatment group (ie, semaglutide [0·05 mg, 0·1 mg, 0·2 mg, 0·3 mg, or 0·4 mg; initiated at 0·05 mg per day and incrementally escalated every 4 weeks] or liraglutide [3·0 mg; initiated at 0·6 mg per day and escalated by 0·6 mg per week]) or matching placebo group (equal injection volume and escalation schedule to active treatment group) using a block size of 56. All treatment doses were delivered once-daily via subcutaneous injections. Participants and investigators were masked to the assigned study treatment but not the target dose. The primary endpoint was percentage weight loss at week 52. The primary analysis was done using intention-to-treat ANCOVA estimation with missing data derived from the placebo pool. This study is registered with ClinicalTrials.gov, number NCT02453711.
Findings: Between Oct 1, 2015, and Feb 11, 2016, 957 individuals were randomly assigned (102-103 participants per active treatment group and 136 in the pooled placebo group). Mean baseline characteristics included age 47 years, bodyweight 111·5 kg, and BMI 39·3 kg/m2. Bodyweight data were available for 891 (93%) of 957 participants at week 52. Estimated mean weight loss was -2·3% for the placebo group versus -6·0% (0·05 mg), -8·6% (0·1 mg), -11·6% (0·2 mg), -11·2% (0·3 mg), and -13·8% (0·4 mg) for the semaglutide groups. All semaglutide groups versus placebo were significant (unadjusted p≤0·0010), and remained significant after adjustment for multiple testing (p≤0·0055). Mean bodyweight reductions for 0·2 mg or more of semaglutide versus liraglutide were all significant (-13·8% to -11·2% vs -7·8%). Estimated weight loss of 10% or more occurred in 10% of participants receiving placebo compared with 37-65% receiving 0·1 mg or more of semaglutide (p<0·0001 vs placebo). All semaglutide doses were generally well tolerated, with no new safety concerns. The most common adverse events were dose-related gastrointestinal symptoms, primarily nausea, as seen previously with GLP-1 receptor agonists.
Interpretation: In combination with dietary and physical activity counselling, semaglutide was well tolerated over 52 weeks and showed clinically relevant weight loss compared with placebo at all doses.
https://sci-hub.se/10.1016/s0140-6736(18)31773-2
Lingvay I, et al. Efficacy and safety of once-weekly semaglutide versus daily canagliflozin as add-on to metformin in patients with type 2 diabetes (SUSTAIN 8): a double-blind, phase 3b, randomised controlled trial. Lancet Diabetes Endocrinol. 2019;7(11):834-44.
Abstract
Background: Existing guidelines for management of type 2 diabetes recommend a patient-centred approach to guide the choice of pharmacological agents. Although glucagon-like peptide-1 (GLP-1) receptor agonists and sodium-glucose cotransporter-2 (SGLT2) inhibitors are increasingly used as second-line agents, direct comparisons between these treatments are insufficient. In the SUSTAIN 8 trial, we compared the efficacy and safety of semaglutide (a GLP-1 receptor agonist) with canagliflozin (an SGLT2 inhibitor) in patients with type 2 diabetes.
Methods: This was a double-blind, parallel-group, phase 3b, randomised controlled trial done at 111 centres in 11 countries. Eligible patients were at least 18 years old and had uncontrolled type 2 diabetes (HbA1c 7·0-10·5% [53-91 mmol/mol]) on stable daily metformin therapy. Patients were randomly assigned (1:1) by use of an interactive web response system to subcutaneous semaglutide 1·0 mg once weekly or oral canagliflozin 300 mg once daily. The primary endpoint was change from baseline in HbA1c, and the confirmatory secondary endpoint was change from baseline in bodyweight, both at week 52. The primary analysis population included all randomly assigned patients, using on-treatment data collected before initiation of rescue medication. The safety analysis was done on a population that included all patients exposed to at least one dose of trial product. The trial was powered for HbA1c and bodyweight superiority under reasonable assumptions. This trial is registered with ClinicalTrials.gov, NCT03136484.
Findings: Between March 15, 2017, and Nov 16, 2018, 788 patients were randomly assigned to semaglutide 1·0 mg (394 patients) or canagliflozin 300 mg (394 patients). 739 patients completed the trial (367 in the semaglutide group and 372 in the canagliflozin group). From overall baseline mean, patients receiving semaglutide had significantly greater reductions in HbA1c and bodyweight than those receiving canagliflozin (HbA1c estimated treatment difference [ETD] -0·49 percentage points, 95% CI -0·65 to -0·33; -5·34 mmol/mol, 95% CI -7·10 to -3·57; p<0·0001; and bodyweight ETD -1·06 kg, 95% CI -1·76 to -0·36; p=0·0029). Gastrointestinal disorders, most commonly nausea, were the most frequently reported adverse events with semaglutide, occurring in 184 (47%) of 392 patients; whereas infections and infestations (defined using the Medical Dictionary for Regulatory Activities, version 21.0), most commonly urinary tract infections, occurred more frequently with canagliflozin, in 136 (35%) of 394 patients. Premature treatment discontinuation because of adverse events occurred in 38 (10%) of 392 patients with semaglutide and in 20 (5%) of 394 patients with canagliflozin. One fatal adverse event confirmed unlikely to be caused by treatment occurred in the semaglutide group.
Interpretation: Once-weekly semaglutide 1·0 mg was superior to daily canagliflozin 300 mg in reducing HbA1c and bodyweight in patients with type 2 diabetes uncontrolled on metformin therapy. These outcomes might guide treatment intensification choices.
https://sci-hub.se/10.1016/s2213-8587(19)30311-0
Pieber TR, et al. Efficacy and safety of oral semaglutide with flexible dose adjustment versus sitagliptin in type 2 diabetes (PIONEER 7): a multicentre, open-label, randomised, phase 3a trial. Lancet Diabetes Endocrinol. 2019;7(7):528-39.
Abstract
Background: Oral semaglutide is the first oral formulation of a glucagon-like peptide-1 (GLP-1) receptor agonist developed for the treatment of type 2 diabetes. We aimed to compare the efficacy and safety of flexible dose adjustments of oral semaglutide with sitagliptin 100 mg.
Methods: In this 52-week, multicentre, randomised, open-label, phase 3a trial, we recruited patients with type 2 diabetes from 81 sites in ten countries. Patients were eligible if they were aged 18 years or older (19 years or older in South Korea), had type 2 diabetes (diagnosed ≥90 days before screening), HbA1c of 7·5-9·5% (58-80 mmol/mol), and were inadequately controlled on stable daily doses of one or two oral glucose-lowering drugs (for 90 days or more before screening). Participants were randomly assigned (1:1) by use of an interactive web-response system, stratified by background glucose-lowering medication at screening, to oral semaglutide with flexible dose adjustments to 3, 7, or 14 mg once daily or sitagliptin 100 mg once daily. To approximate treatment individualisation in clinical practice, oral semaglutide dose could be adjusted on the basis of prespecified HbA1c and tolerability criteria. Two efficacy-related estimands were prespecified: treatment policy (regardless of treatment discontinuation or use of rescue medication) and trial product (on treatment and without use of rescue medication) for participants randomly assigned to treatment. The primary endpoint was achievement of HbA1c of less than 7% (53 mmol/mol) at week 52 and the confirmatory secondary efficacy endpoint was change in bodyweight from baseline to week 52. Safety was assessed in all participants who received at least one dose of study drug. This trial is registered with ClinicalTrials.gov, number NCT02849080, and European Clinical Trials Database, EudraCT number 2015-005593-38, and an open-label extension is ongoing.
Findings: Between Sept 20, 2016, and Feb 7, 2017, of 804 patients assessed for eligibility, 504 were eligible and randomly assigned to oral semaglutide (n=253) or sitagliptin (n=251). Most participants were male (285 [57%] of 504) with a mean age of 57·4 years (SD 9·9). All participants were given at least one dose of their allocated study drug except for one participant in the sitagliptin group. From a mean baseline HbA1c of 8·3% (SD 0·6%; 67 mmol/mol [SD 6·4]), a greater proportion of participants achieved an HbA1c of less than 7% with oral semaglutide than did with sitagliptin (treatment policy estimand: 58% [134 of 230] vs 25% [60 of 238]; and trial product estimand: 63% [123 of 196] vs 28% [52 of 184]). The odds of achieving an HbA1c of less than 7% was significantly better with oral semaglutide than sitagliptin (treatment policy estimand: odds ratio [OR] 4·40, 95% CI 2·89-6·70, p<0·0001; and trial product estimand: 5·54, 3·54-8·68, p<0·0001). The odds of decreasing mean bodyweight from baseline to week 52 were higher with oral semaglutide than with sitagliptin (estimated mean change in bodyweight, treatment policy estimand: -2·6 kg [SE 0·3] vs -0·7 kg [SE 0·2], estimated treatment difference [ETD] -1·9 kg, 95% CI -2·6 to -1·2; p<0·0001; and trial product estimand: -2·9 kg [SE 0·3] vs -0·8 kg [SE 0·3], ETD -2·2 kg, -2·9 to -1·5; p<0·0001). Adverse events occurred in 197 (78%) of 253 participants in the oral semaglutide group versus 172 (69%) of 250 in the sitagliptin group, and nausea was the most common adverse event with oral semaglutide (53 [21%]). Two deaths occurred in the sitagliptin group during the trial.
Interpretation: Oral semaglutide, with flexible dose adjustment, based on efficacy and tolerability, provided superior glycaemic control and weight loss compared with sitagliptin, and with a safety profile consistent with subcutaneous GLP-1 receptor agonists.
https://sci-hub.se/10.1016/s2213-8587(19)30194-9
Mosenzon O, et al. Efficacy and safety of oral semaglutide in patients with type 2 diabetes and moderate renal impairment (PIONEER 5): a placebo-controlled, randomised, phase 3a trial. Lancet Diabetes Endocrinol. 2019;7(7):515-27.
Abstract
Background: Oral semaglutide is the first oral glucagon-like peptide-1 (GLP-1) receptor agonist for glycaemic control in patients with type 2 diabetes. Type 2 diabetes is commonly associated with renal impairment, restricting treatment options. We aimed to investigate the efficacy and safety of oral semaglutide in patients with type 2 diabetes and moderate renal impairment.
Methods: This randomised, double-blind, phase 3a trial was undertaken at 88 sites in eight countries. Patients aged 18 years and older, with type 2 diabetes, an estimated glomerular filtration rate of 30-59 mL/min per 1·73 m2, and who had been receiving a stable dose of metformin or sulfonylurea, or both, or basal insulin with or without metformin for the past 90 days were eligible. Participants were randomly assigned (1:1) by use of an interactive web-response system, with stratification by glucose-lowering medication and renal function, to receive oral semaglutide (dose escalated to 14 mg once daily) or matching placebo for 26 weeks, in addition to background medication. Participants and site staff were masked to assignment. Two efficacy-related estimands were defined: treatment policy (regardless of treatment discontinuation or rescue medication) and trial product (on treatment without rescue medication) in all participants randomly assigned. Endpoints were change from baseline to week 26 in HbA1c (primary endpoint) and bodyweight (confirmatory secondary endpoint), assessed in all participants with sufficient data. Safety was assessed in all participants who received at least one dose of study drug. This trial is registered on ClinicalTrials.gov, number NCT02827708, and the European Clinical Trials Registry, number EudraCT 2015-005326-19, and is now complete.
Findings: Between Sept 20, 2016, and Sept 29, 2017, of 721 patients screened, 324 were eligible and randomly assigned to oral semaglutide (n=163) or placebo (n=161). Mean age at baseline was 70 years (SD 8), and 168 (52%) of participants were female. 133 (82%) participants in the oral semaglutide group and 141 (88%) in the placebo group completed 26 weeks on treatment. At 26 weeks, oral semaglutide was superior to placebo in decreasing HbA1c (estimated mean change of -1·0 percentage point (SE 0·1; -11 mmol/mol [SE 0·8]) vs -0·2 percentage points (SE 0·1; -2 mmol/mol [SE 0·8]); estimated treatment difference [ETD]: -0·8 percentage points, 95% CI -1·0 to -0·6; p<0·0001) and bodyweight (estimated mean change of -3·4 kg [SE 0·3] vs -0·9 kg [SE 0·3]; ETD, -2·5, 95% CI -3·2 to -1·8; p<0·0001) by the treatment policy estimand. Significant differences were seen for the trial product estimand: mean change in HbA1c -1·1 percentage points (SE 0·1; -12 mmol/mol [SE 0·8] versus -0·1 percentage points (SE 0·1; -1 mmol/mol [SE 0·8]; ETD -1·0 percentage points, 95% CI -1·2 to -0·8; p<0·0001); mean change in bodyweight -3·7 kg (SE 0·3) versus -1·1 kg (SE 0·3; ETD -2·7 kg, 95% CI -3·5 to -1·9; p<0·0001). More patients taking oral semaglutide than placebo had adverse events (120 [74%] of 163 vs 105 [65%] of 161), and discontinued treatment as a result (24 [15%] vs eight [5%]). Gastrointestinal events, mainly mild-to-moderate nausea, were more common with oral semaglutide than with placebo. Three deaths occurred during the treatment period that were not condsidered to be treatment related, one in the semaglutide group and two in the placebo group.
Interpretation: Oral semaglutide was effective in patients with type 2 diabetes and moderate renal impairment, potentially providing a new treatment option for this population. Safety, including renal safety, was consistent with the GLP-1 receptor agonist class.
https://sci-hub.se/10.1016/s2213-8587(19)30192-5
Sorli C, et al. Efficacy and safety of once-weekly semaglutide monotherapy versus placebo in patients with type 2 diabetes (SUSTAIN 1): a double-blind, randomised, placebo-controlled, parallel-group, multinational, multicentre phase 3a trial. Lancet Diabetes Endocrinol. 2017;5(4):251-60.
Abstract
Background: Despite a broad range of pharmacological options for the treatment of type 2 diabetes, optimum glycaemic control remains challenging for many patients and new therapies are necessary. Semaglutide is a glucagon-like peptide-1 (GLP-1) analogue in phase 3 development for type 2 diabetes. We assessed the efficacy, safety, and tolerability of semaglutide monotherapy, compared with placebo, in treatment-naive patients with type 2 diabetes who had insufficient glycaemic control with diet and exercise alone.
Methods: We did a double-blind, randomised, parallel-group, international, placebo-controlled phase 3a trial (SUSTAIN 1) at 72 sites in Canada, Italy, Japan, Mexico, Russia, South Africa, UK, and USA (including hospitals, clinical research units, and private offices). Eligible participants were treatment-naive individuals aged 18 years or older with type 2 diabetes treated with only diet and exercise alone for at least 30 days before screening, with a baseline HbA1c of 7·0%-10·0% (53-86 mmol/mol). We randomly assigned participants (2:2:1:1) to either once-weekly subcutaneously injected semaglutide (0·5 mg or 1·0 mg), or volume-matched placebo (0·5 mg or 1·0 mg), for 30 weeks via prefilled PDS290 pen-injectors. Participants did their own injections and were encouraged to administer them on the same day of each week in the same area of their body; the time of day and proximity of meal times was not specified. We did the randomisation with an interactive voice or web response system. Investigators, participants, and the funder of the study remained masked throughout the trial. The primary endpoint was the change in mean HbA1c from baseline to week 30, and the confirmatory secondary endpoint was the change in mean bodyweight from baseline to week 30. We assessed efficacy and safety in the modified intention-to-treat population (ie, all participants who were exposed to at least one dose of study drug); both placebo groups were pooled for assessment. This trial was registered with ClinicalTrials.gov, number NCT02054897.
Findings: Between February 3, 2014, and August 21, 2014, we randomly assigned 388 participants to treatment; 387 received at least one dose of study medication (128 0·5 mg semaglutide, 130 1·0 mg semaglutide, 129 placebo). 17 (13%) of those assigned to 0·5 mg semaglutide, 16 (12%) assigned to 1·0 mg semaglutide, and 14 (11%) assigned to placebo discontinued treatment; the main reason for discontinuation was gastrointestinal adverse events such as nausea. Mean baseline HbA1c was 8·05% (SD 0·85); at week 30, HbA1c significantly decreased by 1·45% (95% CI -1·65 to -1·26) with 0·5 mg semaglutide (estimated treatment difference vs placebo -1·43%, 95% CI -1·71 to -1·15; p<0·0001), significantly decreased by 1·55% (-1·74 to -1·36) with 1·0 mg semaglutide (estimated treatment difference vs placebo -1·53%, -1·81 to -1·25; p<0·0001), and non-significantly decreased by 0·02% (-0·23 to 0·18) with placebo. Mean baseline bodyweight was 91·93 kg (SD 23·83); at week 30, bodyweight significantly decreased by 3·73 kg (95% CI -4·54 to -2·91) with 0·5 mg semaglutide (estimated treatment difference vs placebo -2·75 kg, 95% CI -3·92 to -1·58; p<0·0001), significantly decreased by 4·53 kg (-5·34 to -3·72) with 1·0 mg semaglutide (estimated treatment difference vs placebo -3·56 kg, -4·74 to -2·38; p<0·0001), and non-significantly decreased by 0·98 kg (-1·82 to -0·13) with placebo. No deaths were reported in any of the study groups and most reported adverse events were of mild or moderate severity. The most frequently reported adverse events in both semaglutide groups were gastrointestinal in nature: nausea was reported in 26 (20%) who received 0·5 mg semaglutide, 31 (24%) who received 1·0 mg semaglutide, and 10 (8%) who received placebo, and diarrhoea was reported in 16 (13%) who received 0·5 mg semaglutide, 14 (11%) who received 1·0 mg semaglutide, and three (2%) who received placebo.
Interpretation: Semaglutide significantly improved HbA1c and bodyweight in patients with type 2 diabetes compared with placebo, and showed a similar safety profile to currently available GLP-1 receptor agonists, representing a potential treatment option for such patients.
https://sci-hub.se/10.1016/s2213-8587(17)30013-x
Pratley R, et al. Oral semaglutide versus subcutaneous liraglutide and placebo in type 2 diabetes (PIONEER 4): a randomised, double-blind, phase 3a trial. Lancet. 2019;394(10192):39-50.
Abstract
Background: Glucagon-like peptide-1 (GLP-1) receptor agonists are effective treatments for type 2 diabetes, lowering glycated haemoglobin (HbA1c) and weight, but are currently only approved for use as subcutaneous injections. Oral semaglutide, a novel GLP-1 agonist, was compared with subcutaneous liraglutide and placebo in patients with type 2 diabetes.
Methods: In this randomised, double-blind, double-dummy, phase 3a trial, we recruited patients with type 2 diabetes from 100 sites in 12 countries. Eligible patients were aged 18 years or older, with HbA1c of 7·0-9·5% (53-80·3 mmol/mol), on a stable dose of metformin (≥1500 mg or maximum tolerated) with or without a sodium-glucose co-transporter-2 inhibitor. Participants were randomly assigned (2:2:1) with an interactive web-response system and stratified by background glucose-lowering medication and country of origin, to once-daily oral semaglutide (dose escalated to 14 mg), once-daily subcutaneous liraglutide (dose escalated to 1·8 mg), or placebo for 52 weeks. Two estimands were defined: treatment policy (regardless of study drug discontinuation or rescue medication) and trial product (assumed all participants were on study drug without rescue medication) in all participants who were randomly assigned. The treatment policy estimand was the primary estimand. The primary endpoint was change from baseline to week 26 in HbA1c (oral semaglutide superiority vs placebo and non-inferiority [margin: 0·4%] and superiority vs subcutaneous liraglutide) and the confirmatory secondary endpoint was change from baseline to week 26 in bodyweight (oral semaglutide superiority vs placebo and liraglutide). Safety was assessed in all participants who received at least one dose of study drug. This trial is registered on Clinicaltrials.gov, number NCT02863419, and the European Clinical Trials registry, number EudraCT 2015-005210-30.
Findings: Between Aug 10, 2016, and Feb 7, 2017, 950 patients were screened, of whom 711 were eligible and randomly assigned to oral semaglutide (n=285), subcutaneous liraglutide (n=284), or placebo (n=142). 341 (48%) of 711 participants were female and the mean age was 56 years (SD 10). All participants were given at least one dose of study drug, and 277 (97%) participants in the oral semaglutide group, 274 (96%) in the liraglutide group, and 134 (94%) in the placebo group completed the 52-week trial period. Mean change from baseline in HbA1c at week 26 was -1·2% (SE 0·1) with oral semaglutide, -1·1% (SE 0·1) with subcutaneous liraglutide, and -0·2% (SE 0·1) with placebo. Oral semaglutide was non-inferior to subcutaneous liraglutide in decreasing HbA1c (estimated treatment difference [ETD] -0·1%, 95% CI -0·3 to 0·0; p<0·0001) and superior to placebo (ETD -1·1%, -1·2 to -0·9; p<0·0001) by use of the treatment policy estimand. By use of the trial product estimand, oral semaglutide had significantly greater decreases in HbA1c than both subcutaneous liraglutide (ETD -0·2%, 95% CI -0·3 to -0·1; p=0·0056) and placebo (ETD -1·2%, -1·4 to -1·0; p<0·0001) at week 26. Oral semaglutide resulted in superior weight loss (-4·4 kg [SE 0·2]) compared with liraglutide (-3·1 kg [SE 0·2]; ETD -1·2 kg, 95% CI -1·9 to -0·6; p=0·0003) and placebo (-0·5 kg [SE 0·3]; ETD -3·8 kg, -4·7 to -3·0; p<0·0001) at week 26 (treatment policy). By use of the trial product estimand, weight loss at week 26 was significantly greater with oral semaglutide than with subcutaneous liraglutide (-1·5 kg, 95% CI -2·2 to -0·9; p<0·0001) and placebo (ETD -4·0 kg, -4·8 to -3·2; p<0·0001). Adverse events were more frequent with oral semaglutide (n=229 [80%]) and subcutaneous liraglutide (n=211 [74%]) than with placebo (n=95 [67%]).
Interpretation: Oral semaglutide was non-inferior to subcutaneous liraglutide and superior to placebo in decreasing HbA1c, and superior in decreasing bodyweight compared with both liraglutide and placebo at week 26. Safety and tolerability of oral semaglutide were similar to subcutaneous liraglutide. Use of oral semaglutide could potentially lead to earlier initiation of GLP-1 receptor agonist therapy in the diabetes treatment continuum of care.
https://sci-hub.se/10.1016/s0140-6736(19)31271-1
Enebo LB, et al. Safety, tolerability, pharmacokinetics, and pharmacodynamics of concomitant administration of multiple doses of cagrilintide with semaglutide 2·4 mg for weight management: a randomised, controlled, phase 1b trial. Lancet. 2021;397(10286):1736-48.
Abstract
Background: Cagrilintide, a long-acting amylin analogue, and semaglutide 2·4 mg, a glucagon-like peptide-1 analogue, are both being investigated as options for weight management. We aimed to determine the safety, tolerability, pharmacokinetics, and pharmacodynamics of this drug combination.
Methods: In this randomised, placebo-controlled, multiple-ascending dose, phase 1b trial, individuals aged 18-55 years with a body-mass index 27·0-39·9 kg/m2 and who were otherwise healthy were recruited from a single centre in the USA. The trial included six sequential overlapping cohorts, and in each cohort eligible participants were randomly assigned (3:1) to once-weekly subcutaneous cagrilintide (0·16, 0·30, 0·60, 1·2, 2·4, or 4·5 mg) or matched placebo, in combination with once-weekly subcutaneous semaglutide 2·4 mg, without lifestyle interventions. In each cohort, the doses of cagrilintide and semaglutide were co-escalated in 4-week intervals to the desired dose over 16 weeks, participants were treated at the target dose for 4 weeks, and then followed up for 5 weeks. Participants, investigators, and the sponsor were masked to treatment assignment. The primary endpoint was number of treatment-emergent adverse events from baseline to end of follow-up. Secondary pharmacokinetic endpoints assessed from day of last dose (week 19) to end of treatment (week 20) were area under the plasma concentration-time curve from 0 to 168 h (AUC0-168 h) and maximum concentration [Cmax] of cagrilintide and semaglutide; exploratory pharmacokinetic endpoints were half-life, time to Cmax [tmax], plasma clearance, and volume of distribution of cagrilintide and semaglutide; and exploratory pharmacodynamic endpoints were changes in bodyweight, glycaemic parameters, and hormones. Safety, pharmacokinetic, and pharmacodynamic endpoints were assessed in all participants who were exposed to at least one dose of study drug. This study is registered with ClinicalTrials.gov, NCT03600480, and is now complete.
Findings: Between July 25, 2018, and Dec 17, 2019, 285 individuals were screened and 96 were randomly assigned to cagrilintide (0·16-2·4 mg group n=12; 4·5 mg group n=11) or placebo (n=24), in combination with semaglutide 2·4 mg, of whom 95 were exposed to treatment (one patient in 0·60 mg cagrilintide group was not exposed) and included in the safety and full analysis datasets. The mean age was 40·6 years (SD 9·2), 56 (59%) of 95 participants were men and 51 (54%) were Black or African American. Of 566 adverse events reported in 92 participants (69 [97%] of 71 participants assigned to 0·16-4·5 mg cagrilintide and 23 [96%] of 24 assigned to placebo), 207 (37%) were gastrointestinal disorders. Most adverse events were mild to moderate in severity and the proportion of participants with one or more adverse event was similar across treatment groups. Exposure was proportional to cagrilintide dose and did not affect semaglutide exposure or elimination. AUC0-168 h ranged from 926 nmol × h/L to 24 271 nmol × h/L, and Cmax ranged from 6·14 nmol/L to 170 nmol/L with cagrilintide 0·16-4·5 mg. AUC0-168 h ranged from 12 757 nmol × h/L to 15 305 nmol × h/L, and Cmax ranged from 96·4 nmol/L to 120 nmol/L with semaglutide 2·4 mg. Cagrilintide 0·16-4·5 mg had a half-life of 159-195 h, with a median tmax of 24-72 h. Semaglutide 2·4 mg had a half-life of 145-165 h, with a median tmax of 12-24 h. Plasma clearance and volume of distribution for both cagrilintide and semaglutide were similar across treatment groups. At week 20, mean percentage bodyweight reductions were greater with cagrilintide 1·2 and 2·4 mg than with placebo (15·7% [SE 1·6] for cagrilintide 1·2 mg and 17·1% [1·5] for cagrilintide 2·4 mg vs 9·8% [1·2] for pooled placebo cohorts 1-5; estimated treatment difference of -6·0% [95% CI -9·9 to -2·0] for cagrilintide 1·2 mg and -7·4% [-11·2 to -3·5] for cagrilintide 2·4 mg vs pooled placebo), and with cagrilintide 4·5 mg than with matched placebo (15·4% [1·3] vs 8·0% [2·2]; estimated treatment difference -7·4% [-12·8 to -2·1]), all in combination with semaglutide 2·4 mg. Glycaemic parameters improved in all treatment groups, independently of cagrilintide dose. Changes in hormones were similar across treatment groups.
Interpretation: Concomitant treatment with cagrilintide and semaglutide 2·4 mg was well tolerated with an acceptable safety profile. Future larger and longer trials are needed to fully assess the efficacy and safety of this treatment combination.
https://sci-hub.se/10.1016/s0140-6736(21)00845-x
Ahren B, et al. Efficacy and safety of once-weekly semaglutide versus once-daily sitagliptin as an add-on to metformin, thiazolidinediones, or both, in patients with type 2 diabetes (SUSTAIN 2): a 56-week, double-blind, phase 3a, randomised trial. Lancet Diabetes Endocrinol. 2017;5(5):341-54.
Abstract
Background: Semaglutide is a novel glucagon-like peptide-1 (GLP-1) analogue, suitable for once-weekly subcutaneous administration, in development for treatment of type 2 diabetes. We assessed the efficacy and safety of semaglutide versus the dipeptidyl peptidase-4 (DPP-4) inhibitor sitagliptin in patients with type 2 diabetes inadequately controlled on metformin, thiazolidinediones, or both.
Methods: We did a 56-week, phase 3a, randomised, double-blind, double-dummy, active-controlled, parallel-group, multinational, multicentre trial (SUSTAIN 2) at 128 sites in 18 countries. Eligible patients were aged at least 18 years (or at least 20 years in Japan) and diagnosed with type 2 diabetes, with insufficient glycaemic control (HbA1c 7·0-10·5% [53·0-91·0 mmol/mol]) despite stable treatment with metformin, thiazolidinediones, or both. We randomly assigned participants (2:2:1:1) using an interactive voice or web response system to 56 weeks of treatment with subcutaneous semaglutide 0·5 mg once weekly plus oral sitagliptin placebo once daily, subcutaneous semaglutide 1·0 mg once weekly plus oral sitagliptin placebo once daily, oral sitagliptin 100 mg once daily plus subcutaneous semaglutide placebo 0·5 mg once weekly, or oral sitagliptin 100 mg once daily plus subcutaneous semaglutide placebo 1·0 mg once weekly. The two oral sitagliptin 100 mg groups (with semaglutide placebo 0·5 mg and 1·0 mg) were pooled for the analyses. The primary endpoint was change in HbA1c from baseline to week 56, assessed in the modified intention-to-treat population (all randomly assigned participants who received at least one dose of study drug); change in bodyweight from baseline to week 56 was the confirmatory secondary endpoint. Safety endpoints included adverse events and hypoglycaemic episodes. This trial is registered with ClinicalTrials.gov, number NCT01930188.
Findings: Between Dec 2, 2013, and Aug 5, 2015, we randomly assigned 1231 participants; of the 1225 included in the modified intention-to-treat analysis, 409 received semaglutide 0·5 mg, 409 received semaglutide 1·0 mg, and 407 received sitagliptin 100 mg. Mean baseline HbA1c was 8·1% (SD 0·93); at week 56, HbA1c was reduced by 1·3% in the semaglutide 0·5 mg group, 1·6% in the semaglutide 1·0 mg group, and 0·5% with sitagliptin (estimated treatment difference vs sitagliptin -0·77% [95% CI -0·92 to -0·62] with semaglutide 0·5 mg and -1·06% [-1·21 to -0·91] with semaglutide 1·0 mg; p<0·0001 for non-inferiority and for superiority, for both semaglutide doses vs sitagliptin). Mean baseline bodyweight was 89·5 kg (SD 20·3); at week 56, bodyweight reduced by 4·3 kg with semaglutide 0·5 mg, 6·1 kg with semaglutide 1·0 mg, and 1·9 kg with sitagliptin (estimated treatment difference vs sitagliptin -2·35 kg [95% CI -3·06 to -1·63] with semaglutide 0·5 mg and -4·20 kg [-4·91 to -3·49] with semaglutide 1·0 mg; p<0·0001 for superiority, for both semaglutide doses vs sitagliptin). The proportion of patients who discontinued treatment because of adverse events was 33 (8%) for semaglutide 0·5 mg, 39 (10%) for semaglutide 1·0 mg, and 12 (3%) for sitagliptin. The most frequently reported adverse events in both semaglutide groups were gastrointestinal in nature: nausea was reported in 73 (18%) who received semaglutide 0·5 mg, 72 (18%) who received semaglutide 1·0 mg, and 30 (7%) who received placebo, and diarrhoea was reported in 54 (13%) who received semaglutide 0·5 mg, 53 (13%) who received semaglutide 1·0 mg, and 29 (7%) who received placebo. Seven (2%) patients in the semaglutide 0·5 mg group, two (<1%) in the semaglutide 1·0 mg group, and five (1%) in the sitagliptin group had blood-glucose confirmed hypoglycaemia. There were six fatal events (two in the semaglutide 0·5 mg group, one in the semaglutide 1·0 mg group, and three in the sitagliptin group); none were considered likely to be related to the trial drugs.
Interpretation: Once-weekly semaglutide was superior to sitagliptin at improving glycaemic control and reducing bodyweight in participants with type 2 diabetes on metformin, thiazolidinediones, or both, and had a similar safety profile to that of other GLP-1 receptor agonists. Semaglutide seems to be an effective add-on treatment option for this patient population.
https://sci-hub.se/10.1016/s2213-8587(17)30092-x
Davies M, et al. Semaglutide 2·4 mg once a week in adults with overweight or obesity, and type 2 diabetes (STEP 2): a randomised, double-blind, double-dummy, placebo-controlled, phase 3 trial. Lancet. 2021;397(10278):971-84.
Abstract
Background: This trial assessed the efficacy and safety of the GLP-1 analogue once a week subcutaneous semaglutide 2·4 mg versus semaglutide 1·0 mg (the dose approved for diabetes treatment) and placebo for weight management in adults with overweight or obesity, and type 2 diabetes.
Methods: This double-blind, double-dummy, phase 3, superiority study enrolled adults with a body-mass index of at least 27 kg/m2 and glycated haemoglobin 7-10% (53-86 mmol/mol) who had been diagnosed with type 2 diabetes at least 180 days before screening. Patients were recruited from 149 outpatient clinics in 12 countries across Europe, North America, South America, the Middle East, South Africa, and Asia. Patients were randomly allocated (1:1:1) via an interactive web-response system and stratified by background glucose-lowering medication and glycated haemoglobin, to subcutaneous injection of semaglutide 2·4 mg, or semaglutide 1·0 mg, or visually matching placebo, once a week for 68 weeks, plus a lifestyle intervention. Patients, investigators, and those assessing outcomes were masked to group assignment. Coprimary endpoints were percentage change in bodyweight and achievement of weight reduction of at least 5% at 68 weeks for semaglutide 2·4 mg versus placebo, assessed by intention to treat. Safety was assessed in all patients who received at least one dose of study drug. This study is registered with ClinicalTrials.gov, NCT03552757 and is closed to new participants.
Findings: From June 4 to Nov 14, 2018, 1595 patients were screened, of whom 1210 were randomly assigned to semaglutide 2·4 mg (n=404), semaglutide 1·0 mg (n=403), or placebo (n=403) and included in the intention-to-treat analysis. Estimated change in mean bodyweight from baseline to week 68 was -9·6% (SE 0·4) with semaglutide 2·4 mg vs -3·4% (0·4) with placebo. Estimated treatment difference for semaglutide 2·4 mg versus placebo was -6·2 percentage points (95% CI -7·3 to -5·2; p<0·0001). At week 68, more patients on semaglutide 2·4 mg than on placebo achieved weight reductions of at least 5% (267 [68·8%] of 388 vs 107 [28·5%] of 376; odds ratio 4·88, 95% CI 3·58 to 6·64; p<0·0001). Adverse events were more frequent with semaglutide 2·4 mg (in 353 [87·6%] of 403 patients) and 1·0 mg (329 [81·8%] of 402) than with placebo (309 [76·9%] of 402). Gastrointestinal adverse events, which were mostly mild to moderate, were reported in 256 (63·5%) of 403 patients with semaglutide 2·4 mg, 231 (57·5%) of 402 with semaglutide 1·0 mg, and 138 (34·3%) of 402 with placebo.
Interpretation: In adults with overweight or obesity, and type 2 diabetes, semaglutide 2·4 mg once a week achieved a superior and clinically meaningful decrease in bodyweight compared with placebo.
Level 2
Bettge K, et al. Occurrence of nausea, vomiting and diarrhoea reported as adverse events in clinical trials studying glucagon-like peptide-1 receptor agonists: A systematic analysis of published clinical trials. Diabetes Obes Metab. 2017;19(3):336-47.
Abstract
Aim: GLP-1 receptor agonists (RAs) may cause nausea, vomiting or diarrhoea. The aim of this study was to assess the risk of adverse events (AEs) with GLP-1 RAs and their relation to dose, background medication and duration of action.
Research design and methods: The PubMed database was searched and 32 clinical trials with GLP-1 RAs (phase 3) were selected. We performed a systematic analysis and compared the proportion of patients reporting nausea, vomiting or diarrhoea, for different doses and glucose-lowering background medications, and relative to a reference compound within the subclasses of short- (exenatide b.i.d.) and long-acting (liraglutide) GLP-1 RAs, calculating the relative risks ± 95% confidence intervals.
Results: The risk of nausea was dose-dependent for long-acting (P = .0063) and across ll GLP-1 RAs (P = .0017), and a similar trend was observed for vomiting (P = .23). Diarrhoea was dose-dependent (P = .031). Background treatment with metformin was associated with more nausea (P = .04) and vomiting (P = .0009). Compared to exenatide b.i.d., there was less nausea and diarrhoea with lixisenatide. Compared to liraglutide, there was a similar risk associated with dulaglutide, and less with exenatide q.w. and albiglutide. Long-acting GLP-1 RAs were associated with less nausea and vomiting, but with more diarrhoea than short-acting agents.
Conclusions: GLP-1 RAs are associated with gastrointestinal AEs that are related to dose and background medications (especially metformin) and may vary in a compound-specific manner. Long-acting agents are associated with less nausea and vomiting but with more diarrhoea.
Level 3
McCrimmon RJ, et al. Effects of once-weekly semaglutide vs once-daily canagliflozin on body composition in type 2 diabetes: a substudy of the SUSTAIN 8 randomised controlled clinical trial. Diabetologia. 2020;63(3):473-85.
Abstract
Aims/hypothesis: Intra-abdominal or visceral obesity is associated with insulin resistance and an increased risk for cardiovascular disease. This study aimed to compare the effects of semaglutide 1.0 mg and canagliflozin 300 mg on body composition in a subset of participants from the SUSTAIN 8 Phase IIIB, randomised double-blind trial who underwent whole-body dual-energy x-ray absorptiometry (DXA) scanning.
Methods: Adults (age ≥18 years) with type 2 diabetes, HbA1c 53-91 mmol/mol (7.0-10.5%), on a stable daily dose of metformin (≥1500 mg or maximum tolerated dose) and with an eGFR ≥60 ml min-1 [1.73 m]-2 were randomised 1:1 to semaglutide 1.0 mg once weekly and canagliflozin placebo once daily, or canagliflozin 300 mg once daily and semaglutide placebo once weekly. Body composition was assessed using whole-body DXA scans. The study participants and investigator remained blinded throughout the trial, and quality of DXA scans was evaluated in a blinded manner. Change from baseline to week 52 in total fat mass (kg) was the confirmatory efficacy endpoint.
Results: A subset of 178 participants (semaglutide, n = 88; canagliflozin, n = 90) underwent DXA scanning at screening and were randomised into the substudy. Of these, 114 (semaglutide, n = 53; canagliflozin, n = 61) participants had observed end-of-treatment data included in the confirmatory efficacy analysis. Of the 178 participants in the substudy, numerical improvements in body composition (including fat mass, lean mass and visceral fat mass) were observed after 52 weeks with both treatments. Total fat mass (baseline 33.2 kg) was reduced by 3.4 kg and 2.6 kg with semaglutide and canagliflozin, respectively (estimated treatment difference: -0.79 [95% CI -2.10, 0.51]). Although total lean mass (baseline 51.3 kg) was also reduced by 2.3 kg and 1.5 kg with semaglutide and canagliflozin, respectively (estimated treatment difference: -0.78 [-1.61, 0.04]), the proportion of lean mass (baseline 59.4%) increased by 1.2%- and 1.1%-point, respectively (estimated treatment difference 0.14 [-0.89, 1.17]). Changes in visceral fat mass and overall changes in body composition (assessed by the fat to lean mass ratio) were comparable between the two treatment groups.
Conclusions/interpretation: In individuals with uncontrolled type 2 diabetes on stable-dose metformin therapy, the changes in body composition with semaglutide and canagliflozin were not significantly different. Although numerical improvements in body composition were observed following treatment in both treatment arms, the specific impact of both treatments on body composition in the absence of a placebo arm is speculative at this stage.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6997246/pdf/125_2019_Article_5065.pdf
Frias JP, et al. Tirzepatide versus Semaglutide Once Weekly in Patients with Type 2 Diabetes. N Eng J Med. 2021;385(6):503-15.
Abstract
Background: Tirzepatide is a dual glucose-dependent insulinotropic polypeptide and glucagon-like peptide-1 (GLP-1) receptor agonist that is under development for the treatment of type 2 diabetes. The efficacy and safety of once-weekly tirzepatide as compared with semaglutide, a selective GLP-1 receptor agonist, are unknown.
Methods: In an open-label, 40-week, phase 3 trial, we randomly assigned 1879 patients, in a 1:1:1:1 ratio, to receive tirzepatide at a dose of 5 mg, 10 mg, or 15 mg or semaglutide at a dose of 1 mg. At baseline, the mean glycated hemoglobin level was 8.28%, the mean age 56.6 years, and the mean weight 93.7 kg. The primary end point was the change in the glycated hemoglobin level from baseline to 40 weeks.
Results: The estimated mean change from baseline in the glycated hemoglobin level was -2.01 percentage points, -2.24 percentage points, and -2.30 percentage points with 5 mg, 10 mg, and 15 mg of tirzepatide, respectively, and -1.86 percentage points with semaglutide; the estimated differences between the 5-mg, 10-mg, and 15-mg tirzepatide groups and the semaglutide group were -0.15 percentage points (95% confidence interval [CI], -0.28 to -0.03; P = 0.02), -0.39 percentage points (95% CI, -0.51 to -0.26; P<0.001), and -0.45 percentage points (95% CI, -0.57 to -0.32; P<0.001), respectively. Tirzepatide at all doses was noninferior and superior to semaglutide. Reductions in body weight were greater with tirzepatide than with semaglutide (least-squares mean estimated treatment difference, -1.9 kg, -3.6 kg, and -5.5 kg, respectively; P<0.001 for all comparisons). The most common adverse events were gastrointestinal and were primarily mild to moderate in severity in the tirzepatide and semaglutide groups (nausea, 17 to 22% and 18%; diarrhea, 13 to 16% and 12%; and vomiting, 6 to 10% and 8%, respectively). Of the patients who received tirzepatide, hypoglycemia (blood glucose level, <54 mg per deciliter) was reported in 0.6% (5-mg group), 0.2% (10-mg group), and 1.7% (15-mg group); hypoglycemia was reported in 0.4% of those who received semaglutide. Serious adverse events were reported in 5 to 7% of the patients who received tirzepatide and in 3% of those who received semaglutide.
Conclusions: In patients with type 2 diabetes, tirzepatide was noninferior and superior to semaglutide with respect to the mean change in the glycated hemoglobin level from baseline to 40 weeks. (Funded by Eli Lilly; SURPASS-2 ClinicalTrials.gov number, NCT03987919.).
Rubino D, et al. Effect of Continued Weekly Subcutaneous Semaglutide vs Placebo on Weight Loss Maintenance in Adults With Overweight or Obesity: The STEP 4 Randomized Clinical Trial. JAMA. 2021;35(14):1-12.
Abstract
Importance: The effect of continuing vs withdrawing treatment with semaglutide, a glucagon-like peptide 1 receptor agonist, on weight loss maintenance in people with overweight or obesity is unknown.
Objective: To compare continued once-weekly treatment with subcutaneous semaglutide, 2.4 mg, with switch to placebo for weight maintenance (both with lifestyle intervention) in adults with overweight or obesity after a 20-week run-in with subcutaneous semaglutide titrated to 2.4 mg weekly.
Design, Setting, and Participants: Randomized, double-blind, 68-week phase 3a withdrawal study conducted at 73 sites in 10 countries from June 2018 to March 2020 in adults with body mass index of at least 30 (or ≥27 with ≥1 weight-related comorbidity) and without diabetes.
Interventions: A total of 902 participants received once-weekly subcutaneous semaglutide during run-in. After 20 weeks (16 weeks of dose escalation; 4 weeks of maintenance dose), 803 participants (89.0%) who reached the 2.4-mg/wk semaglutide maintenance dose were randomized (2:1) to 48 weeks of continued subcutaneous semaglutide (n = 535) or switched to placebo (n = 268), plus lifestyle intervention in both groups.
Main Outcomes and Measures: The primary end point was percent change in body weight from week 20 to week 68; confirmatory secondary end points were changes in waist circumference, systolic blood pressure, and physical functioning (assessed using the Short Form 36 Version 2 Health Survey, Acute Version [SF-36]).
Results: Among 803 study participants who completed the 20-week run-in period (with a mean weight loss of 10.6%) and were randomized (mean age, 46 [SD, 12] years; 634 [79%] women; mean body weight, 107.2 kg [SD, 22.7 kg]), 787 participants (98.0%) completed the trial and 741 (92.3%) completed treatment. With continued semaglutide, mean body weight change from week 20 to week 68 was −7.9% vs +6.9% with the switch to placebo (difference, −14.8 [95% CI, −16.0 to −13.5] percentage points; P < .001). Waist circumference (−9.7 cm [95% CI, −10.9 to −8.5 cm]), systolic blood pressure (−3.9 mm Hg [95% CI, −5.8 to −2.0 mm Hg]), and SF-36 physical functioning score (2.5 [95% CI, 1.6-3.3]) also improved with continued subcutaneous semaglutide vs placebo (all P < .001). Gastrointestinal events were reported in 49.1% of participants who continued subcutaneous semaglutide vs 26.1% with placebo; similar proportions discontinued treatment because of adverse events with continued semaglutide (2.4%) and placebo (2.2%).
Conclusions and Relevance: Among adults with overweight or obesity who completed a 20-week run-in period with subcutaneous semaglutide, 2.4 mg once weekly, maintaining treatment with semaglutide compared with switching to placebo resulted in continued weight loss over the following 48 weeks.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7988425/
Wadden TA, et al. Effect of Subcutaneous Semaglutide vs Placebo as an Adjunct to Intensive Behavioral Therapy on Body Weight in Adults With Overweight or Obesity: The STEP 3 Randomized Clinical Trial. JAMA. 2021;325(14):1-11.
Abstract
Importance: Weight loss improves cardiometabolic risk factors in people with overweight or obesity. Intensive lifestyle intervention and pharmacotherapy are the most effective noninvasive weight loss approaches.
Objective: To compare the effects of once-weekly subcutaneous semaglutide, 2.4 mg vs placebo for weight management as an adjunct to intensive behavioral therapy with initial low-calorie diet in adults with overweight or obesity.
Design, Setting, and Participants: Randomized, double-blind, parallel-group, 68-week, phase 3a study (STEP 3) conducted at 41 sites in the US from August 2018 to April 2020 in adults without diabetes (N = 611) and with either overweight (body mass index ≥27) plus at least 1 comorbidity or obesity (body mass index ≥30).
Interventions: Participants were randomized (2:1) to semaglutide, 2.4 mg (n = 407) or placebo (n = 204), both combined with a low-calorie diet for the first 8 weeks and intensive behavioral therapy (ie, 30 counseling visits) during 68 weeks.
Main Outcomes and Measures: The co–primary end points were percentage change in body weight and the loss of 5% or more of baseline weight by week 68. Confirmatory secondary end points included losses of at least 10% or 15% of baseline weight.
Results: Of 611 randomized participants (495 women [81.0%], mean age 46 years [SD, 13], body weight 105.8 kg [SD, 22.9], and body mass index 38.0 [SD, 6.7]), 567 (92.8%) completed the trial, and 505 (82.7%) were receiving treatment at trial end. At week 68, the estimated mean body weight change from baseline was –16.0% for semaglutide vs –5.7% for placebo (difference, −10.3 percentage points [95% CI, −12.0 to −8.6]; P < .001). More participants treated with semaglutide vs placebo lost at least 5% of baseline body weight (86.6% vs 47.6%, respectively; P < .001). A higher proportion of participants in the semaglutide vs placebo group achieved weight losses of at least 10% or 15% (75.3% vs 27.0% and 55.8% vs 13.2%, respectively; P < .001). Gastrointestinal adverse events were more frequent with semaglutide (82.8%) vs placebo (63.2%). Treatment was discontinued owing to these events in 3.4% of semaglutide participants vs 0% of placebo participants.
Conclusions and Relevance: Among adults with overweight or obesity, once-weekly subcutaneous semaglutide compared with placebo, used as an adjunct to intensive behavioral therapy and initial low-calorie diet, resulted in significantly greater weight loss during 68 weeks. Further research is needed to assess the durability of these findings.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7905697/
Davies M, et al. Effect of Oral Semaglutide Compared With Placebo and Subcutaneous Semaglutide on Glycemic Control in Patients With Type 2 Diabetes: A Randomized Clinical Trial. JAMA. 2017;318(15):1460-70.
Abstract
Importance: Glucagon-like peptide-1 (GLP-1) receptor agonists are effective therapies for the treatment of type 2 diabetes and are all currently available as an injection.
Objectives: To compare the effects of oral semaglutide with placebo (primary) and open-label subcutaneous semaglutide (secondary) on glycemic control in patients with type 2 diabetes.
Design, setting, and patients: Phase 2, randomized, parallel-group, dosage-finding, 26-week trial with 5-week follow-up at 100 sites (hospital clinics, general practices, and clinical research centers) in 14 countries conducted between December 2013 and December 2014. Of 1106 participants assessed, 632 with type 2 diabetes and insufficient glycemic control using diet and exercise alone or a stable dose of metformin were randomized. Randomization was stratified by metformin use.
Interventions: Once-daily oral semaglutide of 2.5 mg (n = 70), 5 mg (n = 70), 10 mg (n = 70), 20 mg (n = 70), 40-mg 4-week dose escalation (standard escalation; n = 71), 40-mg 8-week dose escalation (slow escalation; n = 70), 40-mg 2-week dose escalation (fast escalation, n = 70), oral placebo (n = 71; double-blind) or once-weekly subcutaneous semaglutide of 1.0 mg (n = 70) for 26 weeks.
Main outcomes and measures: The primary end point was change in hemoglobin A1c (HbA1c) from baseline to week 26. Secondary end points included change from baseline in body weight and adverse events.
Results: Baseline characteristics were comparable across treatment groups. Of the 632 randomized patients (mean age, 57.1 years [SD, 10.6]; men, 395 (62.7%); diabetes duration, 6.3 years [SD, 5.2]; body weight, 92.3 kg [SD, 16.8]; BMI, 31.7 [SD, 4.3]), 583 (92%) completed the trial. Mean change in HbA1c level from baseline to week 26 decreased with oral semaglutide (dosage-dependent range, -0.7% to -1.9%) and subcutaneous semaglutide (-1.9%) and placebo (-0.3%); oral semaglutide reductions were significant vs placebo (dosage-dependent estimated treatment difference [ETD] range for oral semaglutide vs placebo, -0.4% to -1.6%; P = .01 for 2.5 mg, <.001 for all other dosages). Reductions in body weight were greater with oral semaglutide (dosage-dependent range, -2.1 kg to -6.9 kg) and subcutaneous semaglutide (-6.4 kg) vs placebo (-1.2 kg), and significant for oral semaglutide dosages of 10 mg or more vs placebo (dosage-dependent ETD range, -0.9 to -5.7 kg; P < .001). Adverse events were reported by 63% to 86% (371 of 490 patients) in the oral semaglutide groups, 81% (56 of 69 patients) in the subcutaneous semaglutide group, and 68% (48 of 71 patients) in the placebo group; mild to moderate gastrointestinal events were most common.
Conclusions and relevance: Among patients with type 2 diabetes, oral semaglutide resulted in better glycemic control than placebo over 26 weeks. These findings support phase 3 studies to assess longer-term and clinical outcomes, as well as safety.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5817971/
Friedrichsen M, et al. The effect of semaglutide 2.4 mg once weekly on energy intake, appetite, control of eating, and gastric emptying in adults with obesity. Diabetes Obes MEtab. 2021;23(3):754-62.
Abstract
Aim: To investigate the effects of once-weekly subcutaneous (s.c.) semaglutide 2.4 mg on gastric emptying, appetite, and energy intake in adults with obesity.
Materials and methods: A double-blind, parallel-group trial was conducted in 72 adults with obesity, randomized to once-weekly s.c. semaglutide (dose-escalated to 2.4 mg) or placebo for 20 weeks. Gastric emptying was assessed using paracetamol absorption following a standardized breakfast. Participant-reported appetite ratings and Control of Eating Questionnaire (CoEQ) responses were assessed, and energy intake was measured during ad libitum lunch.
Results: The area under the concentration-time curve (AUC) for paracetamol 0 to 5 hours after a standardized meal (AUC0-5h,para ; primary endpoint) was increased by 8% (P = 0.005) with semaglutide 2.4 mg versus placebo at week 20 (non-significant when corrected for week 20 body weight; P = 0.12). No effect was seen on AUC0-1h,para , maximum observed paracetamol concentration, or time to maximum observed paracetamol concentration. Ad libitum energy intake was 35% lower with semaglutide versus placebo (1736 versus 2676 kJ; estimated treatment difference -940 kJ; P <0.0001). Semaglutide reduced hunger and prospective food consumption, and increased fullness and satiety when compared with placebo (all P <0.02). The CoEQ indicated better control of eating and fewer/weaker food cravings with semaglutide versus placebo (P <0.05). Body weight was reduced by 9.9% with semaglutide and 0.4% with placebo. Safety was consistent with the known profile of semaglutide.
Conclusions: In adults with obesity, once-weekly s.c. semaglutide 2.4 mg suppressed appetite, improved control of eating, and reduced food cravings, ad libitum energy intake and body weight versus placebo. There was no evidence of delayed gastric emptying at week 20, assessed indirectly via paracetamol absorption.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7898914/pdf/DOM-23-754.pdf
Kushner RF, et al. Semaglutide 2.4 mg for the Treatment of Obesity: Key Elements of the STEP Trials 1 to 5. Obesity (Silver Spring). 2020;28(6):1050-61.
Abstract
Objective: The obesity epidemic is a public health concern, warranting further research into pharmacological treatments for weight management (WM) as an adjunct to lifestyle interventions. The Semaglutide Treatment Effect in People with obesity (STEP) program aims to investigate the effect of semaglutide versus placebo on weight loss, safety, and tolerability in adults with obesity or overweight.
Methods: Across five phase 3 trials (NCT03548935, WM; NCT03552757, WM in type 2 diabetes; NCT03611582, WM with intensive behavioral therapy; NCT03548987, sustained WM; and NCT03693430, long-term WM), ~5,000 participants are being randomly assigned to receive semaglutide 2.4 mg once weekly subcutaneously versus placebo. Results will be available in 2020/2021. For all trials, the primary end point is change from baseline to end of treatment in body weight.
Results: Participants have a mean age of 46.2 to 55.3 years, are mostly female (mean: 74.1%-81.0%), and have a mean BMI of 35.7 to 38.5 kg/m2 and a mean waist circumference of 113.0 to 115.7 cm.
Conclusions: The STEP program evaluates the efficacy and safety of semaglutide 2.4 mg subcutaneously once weekly in a broad population. The trials will provide insights on WM in people with obesity with and without type 2 diabetes and on long-term follow-up.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7318657/pdf/OBY-28-1050.pdf
Beck DE, et al. Prospective, randomized, controlled, proof-of-concept study of the Ghrelin mimetic ipamorelin for the management of postoperative ileus in bowel resection patients. Int J Colorectal Dis. 2014;29:1527-34.
Abstract
Background Postoperative ileus is a significant clinical challenge lacking effective management strategies. Ghrelin receptor stimulation has promotility effects in the upper and lower gastrointestinal tract.
Objective This proof-of-concept, phase 2, randomized study evaluated the safety and efficacy of the ghrelin-receptor agonist ipamorelin in the treatment of postoperative ileus following abdominal surgery (ClinicalTrials.gov NCT00672074).
Design The design was a multicenter, double-blind, placebo controlled, clinical trial.
Settings The settings include hospital inpatients. Patients The patients were adults undergoing small and large bowel resection by open or laparoscopic surgery.
Intervention The intervention was intravenous infusions of 0.03-mg/kg ipamorelin vs placebo twice daily, on postoperative day 1 to 7 or hospital discharge. Main outcome measures Safety was assessed by monitoring adverse events and laboratory tests. The key efficacy endpoint was time from first dose of study drug to tolerance of a standardized solid meal.
Results One hundred seventeen patients were enrolled, of whom 114 patients composed the safety and modified intent-to-treat populations. Demographic and disease characteristics were balanced between groups. Overall incidence of any treatment-emergent adverse events was 87.5 % in the ipamorelin group and 94.8 % in placebo group. Median time to first tolerated meal was 25.3 and 32.6 h in the ipamorelin and placebo groups, respectively (p=0.15).
Limitations This proof-of-concept study was small and enrolled patients with a broad range of underlying conditions.
Conclusions Ipamorelin 0.03-mg/kg twice daily for up to 7 days was well tolerated. There were no significant differences between ipamorelin and placebo in the key and secondary efficacy analyses.
https://sci-hub.se/10.1007/s00384-014-2030-8
Faillie JL, et al. Association of Bile Duct and Gallbladder Diseases With the Use of Incretin-Based Drugs in Patients With Type 2 Diabetes Mellitus. JAMA Intern Med. 2016;176(10):1474-81.
Abstract
Importance: The use of dipeptidyl-peptidase-4 (DPP-4) inhibitors and glucagon-like peptide 1 (GLP-1) analogues-a group of drugs used in the management of type 2 diabetes mellitus-may be associated with an increased risk of bile duct and gallbladder disease. To date, no observational study has assessed this possible association.
Objective: To determine whether the use of DPP-4 inhibitors and GLP-1 analogues is associated with an increased risk of incident bile duct and gallbladder disease in patients with type 2 diabetes.
Design, setting, and participants: A population-based cohort study linked the United Kingdom Clinical Practice Research Datalink with the Hospital Episodes Statistics database, yielding a cohort of 71 369 patients, 18 years or older, initiating an antidiabetic drug (including oral and injectable agents) between January 1, 2007, and March 31, 2014.
Exposures: Current use of DPP-4 inhibitors and GLP-1 analogues (alone or in combination therapy) compared with current use of at least 2 oral antidiabetic drugs.
Main outcomes and measures: Time-dependent Cox proportional hazards models were used to estimate hazard ratios (HRs) with 95% CIs of incident bile duct or gallbladder events (cholelithiasis, cholecystitis, cholangitis) causing hospitalization, comparing current use of DPP-4 inhibitors and GLP-1 analogues with current use of at least 2 oral antidiabetic drugs.
Results: During 227 994 person-years of follow-up, 853 of the 71 369 patients were hospitalized for bile duct and gallbladder disease (incidence rate per 1000 person-years, 3.7; 95% CI, 3.5-4.0). Current use of DPP-4 inhibitors was not associated with an increased risk of bile duct and gallbladder disease compared with current use of at least 2 oral antidiabetic drugs (3.6 vs 3.3 per 1000 person-years; adjusted HR, 0.99; 95% CI, 0.75-1.32). In contrast, the use of GLP-1 analogues was associated with an increased risk of bile duct and gallbladder disease compared with current use of at least 2 oral antidiabetic drugs (6.1 vs 3.3 per 1000 person-years; adjusted HR, 1.79; 95% CI, 1.21-2.67). In a secondary analysis, GLP-1 analogues were also associated with an increased risk of cholecystectomy (adjusted HR, 2.08; 95% CI, 1.08-4.02).
Conclusions and relevance: The use of GLP-1 analogues was associated with an increased risk of bile duct and gallbladder disease. Physicians should be aware of this potential adverse event when prescribing these drugs.
https://sci-hub.se/10.1001/jamainternmed.2016.1531
Buckley ST, et al. Transcellular stomach absorption of a derivatized glucagon-like peptide-1 receptor agonist. Sci Transl Med. 2018;10(467):eaar7047.
Abstract
Oral administration of therapeutic peptides is hindered by poor absorption across the gastrointestinal barrier and extensive degradation by proteolytic enzymes. Here, we investigated the absorption of orally delivered semaglutide, a glucagon-like peptide-1 analog, coformulated with the absorption enhancer sodium N-[8-(2-hydroxybenzoyl) aminocaprylate] (SNAC) in a tablet. In contrast to intestinal absorption usually seen with small molecules, clinical and preclinical dog studies revealed that absorption of semaglutide takes place in the stomach, is confined to an area in close proximity to the tablet surface, and requires coformulation with SNAC. SNAC protects against enzymatic degradation via local buffering actions and only transiently enhances absorption. The mechanism of absorption is shown to be compound specific, transcellular, and without any evidence of effect on tight junctions. These data have implications for understanding how highly efficacious and specific therapeutic peptides could be transformed from injectable to tablet-based oral therapies.
Level 7
Li Q, et al. Semaglutide attenuates excessive exercise-induced myocardial injury through inhibiting oxidative stress and inflammation in rats. Life Sci. 2020;250:117531.
Abstract
Aims: To investigate the protective effects and mechanism of semaglutide on exercise-induced myocardial injury.
Main methods: Effects of semaglutide on lipopolysaccharide (LPS)-induced oxidative stress injuries and inflammatory response were assessed in H9c2 cell via MTT assay and Western blot. Quiet control group, over training group and three doses of semaglutide treated overtraining groups were subjected to the swimming training with increasing load for consecutive 10 weeks. Immediately after the last training, the body weight, myocardial morphological changes, injury markers and inflammatory response related proteins of the model rats were analyzed.
Key findings: Semaglutide at three concentrations in LPS treated H9c2 cells significantly increased the survival rate and inhibited the apoptosis of cardiomyocytes. Moreover, semaglutide activated AMPK pathway, improve autophagy and inhibited reactive oxygen species production in LPS treated H9C2 cells. In vivo results further revealed that chronic treatment of semaglutide induced significant increase in myocardial injury markers. The pathological histology analysis results showed that semaglutide ameliorated myocardial morphological changes, reduced area of lipid accumulation and significantly decreased the expression levels of NF-κB, TNF-α and IL-1β.
Significance: Semaglutide exert the protective effects on exercise-induced cardiomyopathy by activating AMPK pathway, increasing autophagy, reducing the production of ROS and inflammation-related proteins.
https://sci-hub.se/10.1016/j.lfs.2020.117531
Gabery S, et al. Semaglutide lowers body weight in rodents via distributed neural pathways. JCI Insight. 2020;5(6):e133429.
Abstract
Semaglutide, a glucagon-like peptide 1 (GLP-1) analog, induces weight loss, lowers glucose levels, and reduces cardiovascular risk in patients with diabetes. Mechanistic preclinical studies suggest weight loss is mediated through GLP-1 receptors (GLP-1Rs) in the brain. The findings presented here show that semaglutide modulated food preference, reduced food intake, and caused weight loss without decreasing energy expenditure. Semaglutide directly accessed the brainstem, septal nucleus, and hypothalamus but did not cross the blood-brain barrier; it interacted with the brain through the circumventricular organs and several select sites adjacent to the ventricles. Semaglutide induced central c-Fos activation in 10 brain areas, including hindbrain areas directly targeted by semaglutide, and secondary areas without direct GLP-1R interaction, such as the lateral parabrachial nucleus. Automated analysis of semaglutide access, c-Fos activity, GLP-1R distribution, and brain connectivity revealed that activation may involve meal termination controlled by neurons in the lateral parabrachial nucleus. Transcriptomic analysis of microdissected brain areas from semaglutide-treated rats showed upregulation of prolactin-releasing hormone and tyrosine hydroxylase in the area postrema. We suggest semaglutide lowers body weight by direct interaction with diverse GLP-1R populations and by directly and indirectly affecting the activity of neural pathways involved in food intake, reward, and energy expenditure.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7213778/pdf/jciinsight-5-133429.pdf
Abdullah DM, et al. Semaglutide early intervention attenuated testicular dysfunction by targeting the GLP-1-PPAR-α-Kisspeptin-Steroidogenesis signaling pathway in a testicular ischemia-reperfusion rat model. Peptides.2022;149:170711.
Abstract
Testicular torsion is a serious emergency and a well-known cause of male infertility. It represents 10 %-15 % of scrotal diseases in children. Kisspeptin (KISS1) is a hormone secreted from the hypothalamic nuclei and testis, but its role in testis is not fully understood. Semaglutide is a novel antidiabetic glucagon-like peptide 1 (GLP-1) analog. Hence, we designed the current study to elucidate the possible ameliorative effect of semaglutide on ischemia/reperfusion-induced testicular dysfunction in rats and highlight the role of the testicular GLP-1/PCG-1α-PPAR-α-KISS1 signaling pathway. We randomly divided 50 male Sprague Dawley into five equal groups (10 rats each): SHAM, exendin 9-39 -treated (EX), testicular torsion/detorsion (T/D), testicular torsion/detorsion and semaglutide-treated (SEM + T/D), and testicular torsion/detorsion, exendin, and semaglutide-treated (EX + SEM + T/D). We quantified serum follicle-stimulating hormone, luteinizing hormone, total testosterone, testicular oxidative stress markers, testicular gene expression of GLP-1/KISS1 pathway-related genes (KISS1, KISS1R, GLP-1, GLP-1R, PGC-1α, PPAR-α), steroidogenesis pathway-related genes (STAR, CYP11A1, CYP17A1, HSD17B3, CYP19A1), HO-1, Nrf-2, and testicular protein expression of HIF-1α, TNF-α, NF-κβ, Caspase-3, FAS, proliferating cell nuclear antigen, and KISS1 through testicular histopathology and immunohistochemistry assays. Testicular torsion/detorsion markedly elevated proapoptotic, proinflammatory, and oxidative stress marker levels, noticeably downregulating the expression of GLP-1/KISS1 and steroidogenesis pathway-related proteins. Semaglutide administration significantly ameliorated all these deleterious effects. Nevertheless, injecting exendin, a GLP1-R antagonist, before semaglutide abolished all the documented improvements. We concluded that semaglutide ameliorated ischemia/reperfusion-induced testicular dysfunction by modulating the GLP-1/PGC-1α-PPAR-α/KISS1/steroidogenesis signaling pathway, improving testicular oxidative state, and suppressing testicular inflammation and apoptosis.
No full text available
Ghidewon M, et al. Growth differentiation factor 15 (GDF15) and semaglutide inhibit food intake and body weight through largely distinct, additive mechanisms. Diabetes Obes Metab. 2022;24(6):1010-20.
Abstract
Aims: To evaluate whether the potent hypophagic and weight-suppressive effects of growth differentiation factor-15 (GDF15) and semaglutide combined would be a more efficacious antiobesity treatment than either treatment alone by examining whether the neural and behavioural mechanisms contributing to their anorectic effects were common or disparate.
Materials/methods: Three mechanisms were investigated to determine how GDF15 and semaglutide induce anorexia: the potentiation of the intake suppression by gastrointestinal satiation signals; the reduction in motivation to feed; and the induction of visceral malaise. We then compared the effects of short-term, combined GDF15 and semaglutide treatment on weight loss to the individual treatments. Rat pharmaco-behavioural experiments assessed whether GDF15 or semaglutide added to the satiating effects of orally gavaged food and exogenous cholecystokinin (CCK). A progressive ratio operant paradigm was used to examine whether GDF15 or semaglutide reduced feeding motivation. Pica behaviour (ie, kaolin intake) and conditioned affective food aversion testing were used to evaluate visceral malaise. Additionally, fibre photometry studies were conducted in agouti-related protein (AgRP)-Cre mice to examine whether GDF15 or semaglutide, alone or in combination with CCK, modulate calcium signalling in hypothalamic AgRP neurons.
Results: Semaglutide reduced food intake by amplifying the feeding-inhibitory effect of CCK or ingested food, inhibited the activity of AgRP neurons when combined with CCK, reduced feeding motivation and induced malaise. GDF15 induced visceral malaise but, strikingly, did not affect feeding motivation, the satiating effect of ingested food or CCK signal processing. Combined GDF15 and semaglutide treatment produced greater food intake and body weight suppression than did either treatment alone, without enhancing malaise.
Conclusions: GDF15 and semaglutide reduce food intake and body weight through largely distinct processes that produce greater weight loss and feeding suppression when combined.
No Full Text Available
Jiang Z, et al. Semaglutide ameliorates lipopolysaccharide-induced acute lung injury through inhibiting HDAC5-mediated activation of NF-κB signaling pathway. Hum Exp Toxicol. 2022;41:9603271221125931.
Abstract
Background: As a life-threatening respiratory syndrome, acute lung injury (ALI) is characterized by uncontrollable inflammatory activities. Semaglutide (SEM) has been identified as an effective anti-inflammatory drug in a variety of diseases. This study intended to explore the functional effect and potential mechanisms of SEM in ALI.
Methods: Lipopolysaccharide (LPS) was used to construct an in vivo ALI model based on Sprague-Dawley (SD) rats and an in vitro ALI model based on human pulmonary artery endothelial cells (HPAECs). Hematoxylin & eosin (H&E) staining and ELISA were applied to evaluate the histopathological changes in pulmonary tissues and detect TNF-α and IL-6 levels. RT-qPCR and Western blotting were used to measure gene and protein expressions in pulmonary tissues and cells. HPAEC viability and apoptosis were evaluated by CCK-8 method and flow cytometry methods.
Results: Semaglutide pretreatment significantly mitigated pulmonary injury, reduced TNF-α and IL-6 production, and led to a decrease in cleaved caspase-3 level and an increase in Bcl-2 level, suggesting SEM could ameliorate LPS-induced ALI in rats. In vitro, SEM increased the proliferative capability and mitigated inflammation and apoptosis in LPS-stimulated HPAECs. In addition, SEM inhibited HDAC5-mediated NF-κB signaling pathway in HPAECs. HDAC5 overexpression or NF-κB signaling activation could partly impair SEM-mediated protective effects against LPS-induced damage to HPAECs.
Conclusion: Semaglutide restrains LPS-induced ALI by inhibiting HDAC5/NF-κB signaling pathway.
https://journals.sagepub.com/doi/epub/10.1177/09603271221125931
Level 8
Coon SA, et al. Semaglutide once-weekly: improved efficacy with a new safety warning. Expert Rev Clin Pharmacol. 2018;11(11):1061-72.
Abstract
Semaglutide once-weekly glucagon-like peptide-1 receptor agonist (GLP-1 RA) injection has been approved as an adjunct to diet and exercise to improve glycemic control in type 2 diabetes mellitus (T2DM). Areas Covered: The safety and efficacy of the semaglutide once-weekly injection are reviewed using results from preliminary pharmacology studies and later-phase randomized control trials (RCTs) and meta-analyses. Semaglutide once-weekly is compared to placebo and active comparators for T2DM in the SUSTAIN clinical trial series, with outcomes of: glycemic control, weight loss, major adverse cardiovascular events, and adverse effects. Risk for diabetic retinopathy complications (DRCs) is reviewed in detail, due to significantly higher risk for DRCs seen in SUSTAIN 6. SUSTAIN 6 is the first instance of a GLP-1 RA demonstrating significantly increased risk for DRCs. Semaglutide’s current regulatory approvals, practice considerations, and cost-effectiveness compared to similar therapies are also considered. Expert Commentary: Semaglutide demonstrates high glycemic efficacy and favorable safety profile, and reduces the risk for cardiovascular events. Mild to moderate gastrointestinal events and retinopathy complications were more common with semaglutide compared to placebo, though serious adverse events were similar to controls and infrequent. Improved clinical efficacy should be carefully weighed against the risk for GI and retinopathy complications.
https://sci-hub.se/10.1080/17512433.2018.1534201
Phillips A, et al. Clinical review of subcutaneous semaglutide for obesity. J Clin Pharm Ther. 2022;47(2):184-93.
Abstract
What is known and objective: The purpose of this review paper is to review the efficacy and safety of subcutaneous semaglutide, marketed as Wegovy, a glucagon-like peptide-1 receptor agonist for obesity management.
Methods: A MEDLINE search (1970 to June 2021) was conducted to identify Phase 3 trials of subcutaneous semaglutide for obesity management. Published Phase 3 trials from The Semaglutide Treatment Effect in People with obesity (STEP) program were reviewed and summarized.
Results and discussion: Based on four Phase 3 trials, subcutaneous semaglutide as 2.4 mg once weekly was compared in efficacy and safety among 5000 randomized participants who were overweight or had obesity. A change in body weight from baseline to end of study was the primary outcome in the STEP program. Participants who received semaglutide had a dose-dependent reduction in body weight from baseline, compared to placebo. Higher percentages of participants had 5%-10% weight reduction from baseline when receiving subcutaneous semaglutide. The patient population was mainly middle-aged female participants with Class II obesity. Additional studies are needed, especially active-comparator trials, to determine the efficacy and safety of semaglutide in a diverse patient population.
What is new and conclusion: Subcutaneous semaglutide is another available option as adjunct therapy to lifestyle modifications for people who are overweight or have obesity based on body weight and body mass index. It resulted in more weight reduction than placebo with gastrointestinal adverse events being the most common safety concerns. Clinical utilization of subcutaneous semaglutide will be determined, as insurance coverage will be a limitation for this new medication.
https://onlinelibrary.wiley.com/doi/epdf/10.1111/jcpt.13574
Tan X, et al. Efficacy and safety of once-weekly semaglutide for the treatment of type 2 diabetes. Expert Opin Investig Drugs. 2017;26(9):1083-89.
Abstract
Type 2 diabetes is a chronic metabolic disease characterized by persistent hyperglycemia resulting from progressive deficient of insulin in patients with a background of insulin resistance. Current treatment algorithms recommended by American Diabetes Association/The European Association for the Study of Diabetes promote a patient-centered approach that takes into account a comprehensive consideration of pharmacological properties of drugs, including glucose-lowering action, effects on body weight, correction on multiple pathophysiologic defects, tolerability, and long-term safety. Glucagon-likepeptide1 (GLP-1) receptor analogues are appealing due to the improved glycemic control in a glucose-dependent manner, modest weight loss and low risk of hypoglycemia. Areas covered: Semaglutide (Novo Nordisk), a once-weekly GLP-1 analogue, is currently in the phase III clinical trial for the treatment of type 2 diabetes. This article aims to review the pharmacological and clinical profiles of semaglutide based on the available clinical data. Expert opinion: Semaglutide achieved greater reduction from baseline in HbA1c in comparison to placebo. The greater proportion of patients in semaglutide group than that in placebo group achieved target HbA1c <7.0% and <6.5%, respectively. Semaglutide is the second GLP-1 analogue contributing to the reduced bodyweight and improving obesity related complications. More importantly, semaglutide is beneficial to diabetic patients with high cardiovascular risk according to the recently completed phase III trial. The incidence of gastrointestinal adverse effects increased with semaglutide dose.
https://sci-hub.se/10.1080/13543784.2017.1360274
Goldenberg RM, et al. Semaglutide: Review and Place in Therapy for Adults With Type 2 Diabetes.Can J Diabetes. 2019;43(2):136-45.
Abstract
Guidelines increasingly highlight the importance of multifactorial management in type 2 diabetes, in contrast to the more traditional focus on glycemic control. Semaglutide, a recently approved glucagon-like peptide-1 receptor agonist, is indicated in Canada for adults with type 2 diabetes to improve glycemic control as monotherapy with diet and exercise when metformin is inappropriate or as an add-on to either metformin alone or metformin plus a sulfonylurea or basal insulin. The Semaglutide Unabated Sustainability in Treatment of Type 2 Diabetes (SUSTAIN) clinical trial program for semaglutide comprises 6 pivotal global phase 3a trials (SUSTAIN 1 through 6) and 2 Japanese phase 3a trials. Phase 3b trials include SUSTAIN 7, and SUSTAIN 8 and 9 (both ongoing). Results from the completed trials support the superiority of semaglutide for reduction of glycated hemoglobin levels and weight loss vs. placebo as well as active comparators, including sitagliptin, exenatide extended-release, dulaglutide and insulin glargine. SUSTAIN 6 trial data confirmed cardiovascular safety and demonstrated significant reductions in major cardiovascular events with semaglutide vs. placebo, an outcome that confirmed the noninferiority of semaglutide. The robust and sustained effects of semaglutide on glycated hemoglobin levels and weight loss vs. comparators, as well as its safety and possible cardiovascular benefit, address an unmet need in the treatment of type 2 diabetes. This article overviews data from across the semaglutide clinical trial program, including efficacy and safety results and findings from post hoc analyses. The potential place of semaglutide in clinical practice is discussed.
https://sci-hub.se/10.1016/j.jcjd.2018.05.008
Lyseng-Williamson KA. Glucagon-Like Peptide-1 Receptor Analogues in Type 2 Diabetes: Their Use and Differential Features. Clin Drug Investig. 2019;39(8):805-19.
Abstract
Glucagon-like peptide-1 receptor agonists (GLP-1RAs) are well established as effective adjuncts to lifestyle modification in the treatment of type 2 diabetes (T2D) as monotherapy or in combination with oral glucose-lowering drugs ± insulin. The six subcutaneous GLP-1RA formulations (i.e. twice-daily exenatide, once-daily liraglutide and lixisenatide, and once-weekly dulaglutide, exenatide and semaglutide) currently available in the EU and USA have many similarities, but also some unique features and properties. By stimulating GLP-1 receptors, GLP-1RAs increase insulin secretion and suppress glucagon release in a glucose-dependent manner, thereby improving clinical and patient-reported outcomes related to glycaemic control and weight. They also have been shown to reduce, or at least not increase, the risk of major cardiovascular outcomes. GLP-1RAs are generally well tolerated, with gastrointestinal and injection-site reactions being the most troublesome drug-related adverse events, and are associated with a very low intrinsic risk of hypoglycaemia. Treatment with GLP-1RAs should be customized to meet the clinical needs and personal preferences of the individual.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6746674/pdf/40261_2019_Article_826.pdf
Gallwitz B, et al. Clinical Perspectives on the Use of Subcutaneous and Oral Formulations of Semaglutide. Front Endocrinol (Laussane). 2021;12:645507.
Abstract
Early and effective glycemic control can prevent or delay the complications associated with type 2 diabetes (T2D). The benefits of glucagon-like peptide-1 receptor agonists (GLP-1RAs) are becoming increasingly recognized and they now feature prominently in international T2D treatment recommendations and guidelines across the disease continuum. However, despite providing effective glycemic control, weight loss, and a low risk of hypoglycemia, GLP-1RAs are currently underutilized in clinical practice. The long-acting GLP-1RA, semaglutide, is available for once-weekly injection and in a new once-daily oral formulation. Semaglutide is an advantageous choice for the treatment of T2D since it has greater efficacy in reducing glycated hemoglobin and body weight compared with other GLP-1RAs, has demonstrated benefits in reducing major adverse cardiovascular events, and has a favorable profile in special populations (e.g., patients with hepatic impairment or renal impairment). The oral formulation represents a useful option to help improve acceptance and adherence compared with injectable formulations for patients with a preference for oral therapy, and may lead to earlier and broader use of GLP-1RAs in the T2D treatment trajectory. Oral semaglutide should be taken on an empty stomach, which may influence the choice of formulation. As with most GLP-1RAs, initial dose escalation of semaglutide is required for both formulations to mitigate gastrointestinal adverse events. There are also specific dose instructions to follow with oral semaglutide to ensure sufficient gastric absorption. The evidence base surrounding the clinical use of semaglutide is being further expanded with trials investigating effects on diabetic retinopathy, cardiovascular outcomes, and on the common T2D comorbidities of obesity, chronic kidney disease, and non-alcoholic steatohepatitis. These will provide further information about whether the benefits of semaglutide extend to these other indications.
https://www.frontiersin.org/articles/10.3389/fendo.2021.645507/full
Knudsen LB,e t al. The discovery and development of liraglutide and semaglutide. Front. Endocrinol (Lausanne). 2019;10:155.
Abstract
The discovery of glucagon-like peptide-1 (GLP-1), an incretin hormone with important effects on glycemic control and body weight regulation, led to efforts to extend its half-life and make it therapeutically effective in people with type 2 diabetes (T2D). The development of short- and then long-acting GLP-1 receptor agonists (GLP-1RAs) followed. Our article charts the discovery and development of the long-acting GLP-1 analogs liraglutide and, subsequently, semaglutide. We examine the chemistry employed in designing liraglutide and semaglutide, the human and non-human studies used to investigate their cellular targets and pharmacological effects, and ongoing investigations into new applications and formulations of these drugs. Reversible binding to albumin was used for the systemic protraction of liraglutide and semaglutide, with optimal fatty acid and linker combinations identified to maximize albumin binding while maintaining GLP-1 receptor (GLP-1R) potency. GLP-1RAs mediate their effects via this receptor, which is expressed in the pancreas, gastrointestinal tract, heart, lungs, kidneys, and brain. GLP-1Rs in the pancreas and brain have been shown to account for the respective improvements in glycemic control and body weight that are evident with liraglutide and semaglutide. Both liraglutide and semaglutide also positively affect cardiovascular (CV) outcomes in individuals with T2D, although the precise mechanism is still being explored. Significant weight loss, through an effect to reduce energy intake, led to the approval of liraglutide (3.0 mg) for the treatment of obesity, an indication currently under investigation with semaglutide. Other ongoing investigations with semaglutide include the treatment of non-alcoholic fatty liver disease (NASH) and its use in an oral formulation for the treatment of T2D. In summary, rational design has led to the development of two long-acting GLP-1 analogs, liraglutide and semaglutide, that have made a vast contribution to the management of T2D in terms of improvements in glycemic control, body weight, blood pressure, lipids, beta-cell function, and CV outcomes. Furthermore, the development of an oral formulation for semaglutide may provide individuals with additional benefits in relation to treatment adherence. In addition to T2D, liraglutide is used in the treatment of obesity, while semaglutide is currently under investigation for use in obesity and NASH.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6474072/pdf/fendo-10-00155.pdf
Phillips A, et al. Clinical review of subcutaneous semaglutide for obesity. J Clin Pharm Ther. 2022;47(2):184-93.
Abstract
What is known and objective: The purpose of this review paper is to review the efficacy and safety of subcutaneous semaglutide, marketed as Wegovy, a glucagon-like peptide-1 receptor agonist for obesity management. Methods: A MEDLINE search (1970 to June 2021) was conducted to identify Phase 3 trials of subcutaneous semaglutide for obesity management. Published Phase 3 trials from The Semaglutide Treatment Effect in People with obesity (STEP) program were reviewed and summarized. Results and discussion: Based on four Phase 3 trials, subcutaneous semaglutide as 2.4 mg once weekly was compared in efficacy and safety among 5000 randomized participants who were overweight or had obesity. A change in body weight from baseline to end of study was the primary outcome in the STEP program. Participants who received semaglutide had a dose-dependent reduction in body weight from baseline, compared to placebo. Higher percentages of participants had 5%-10% weight reduction from baseline when receiving subcutaneous semaglutide. The patient population was mainly middle-aged female participants with Class II obesity. Additional studies are needed, especially active-comparator trials, to determine the efficacy and safety of semaglutide in a diverse patient population. What is new and conclusion: Subcutaneous semaglutide is another available option as adjunct therapy to lifestyle modifications for people who are overweight or have obesity based on body weight and body mass index. It resulted in more weight reduction than placebo with gastrointestinal adverse events being the most common safety concerns. Clinical utilization of subcutaneous semaglutide will be determined, as insurance coverage will be a limitation for this new medication.