Level 7
Li B, et al. Neurotrophic peptides incorporating adamantane improve learning and memory, promote neurogenesis and synaptic plasticity in mice. FEBS Lett. 2010;584(15):3359-65.
Abstract
Development of neurotrophic peptidergic drugs that can mimic neurotrophins and promote neurogenesis and maturation of newborn cells into mature functional neurons represents an exciting therapeutic opportunity for treatment of Alzheimer disease and other learning and memory disorders as well as enhancing cognition of normal individuals. Here we report the design of a peptidergic compound, Ac-DGGLAG-NH2, called P21, when administered peripherally, enhanced learning as well as both short-term and spatial reference memories of normal adult C57Bl6 mice. P21 induced enhancement of neurogenesis and maturation of newly born neurons in the granular cell layer and subgranular zone of the dentate gyrus.
https://febs.onlinelibrary.wiley.com/doi/epdf/10.1016/j.febslet.2010.06.025
Carter BD, et al. Differential Regulation of p21ras Activation in Neurons by Nerve Growth Factor and Brain-derived Neurotrophic Factor. JBC. 1995;270(37):21751-57.
Abstract
Neurotrophins activate the Trk tyrosine kinase receptors, which subsequently initiate signaling pathways that have yet to be fully resolved, resulting in neuronal survival and differentiation. The ability of nerve growth factor (NGF) and brain-derived neurotrophic factor (BDNF) to activate GTP binding to p21ras was investigated using cultured embryonic chick neurons. In both sympathetic and sensory neurons, the addition of NGF markedly increased the formation of Ras-GTP. The magnitude of the effect was found to depend upon the developmental stage, peaking at embryonic day 11 in sympathetic neurons and at embryonic day 9 in sensory neurons, times when large numbers of neurons depend on NGF for survival. Surprisingly, following the addition of BDNF, no formation of Ras-GTP could be observed in neurons cultured with BDNF. When sensory neurons were cultured with NGF alone, both NGF and BDNF stimulated GTP binding to Ras. In rat cerebellar granule cells, while the acute exposure of these cells to BDNF resulted in the formation Ras-GTP, no response was observed following previous exposure of the cells to BDNF, as was observed with sensory neurons. However, this desensitization was not observed in a transformed cell line expressing TrkB. In neurons, the mechanism underlying the loss of the BDNF response appeared to involve a dramatic loss of binding to cell-surface receptors, as determined by cross-linking with radiolabeled BDNF. Receptor degradation could not account for the desensitization since cell lysates from neurons pretreated with BDNF revealed that the levels of TrkB were comparable to those in untreated cells. These results indicate that in neurons, the pathways activated by NGF and BDNF are differentially regulated and that prolonged exposure to BDNF results in the inability of TrkB to bind its ligand.
https://sci-hub.se/10.1074/jbc.270.37.21751
Zhao L,et al. Role of p21-activated kinase pathway defects in the cognitive deficits of Alzheimer’s disease. Nature Neurosci. 2006;9(2):232-42.
Abstract
Defects in dendritic spines are common to several forms of cognitive deficits, including mental retardation and Alzheimer disease. Because mutation of p21-activated kinase (PAK) can lead to mental retardation and because PAK-cofilin signaling is critical in dendritic spine morphogenesis and actin dynamics, we hypothesized that the PAK pathway is involved in synaptic and cognitive deficits in Alzheimer disease. Here, we show that PAK and its activity are markedly reduced in Alzheimer disease and that this is accompanied by reduced and redistributed phosphoPAK, prominent cofilin pathology and downstream loss of the spine actinregulatory protein drebrin, which cofilin removes from actin. We found that b-amyloid (Ab) was directly involved in PAK signaling deficits and drebrin loss in Ab oligomer–treated hippocampal neurons and in the Appswe transgenic mouse model bearing a double mutation leading to higher Ab production. In addition, pharmacological PAK inhibition in adult mice was sufficient to cause similar cofilin pathology, drebrin loss and memory impairment, consistent with a potential causal role of PAK defects in cognitive deficits in Alzheimer disease.
https://sci-hub.se/10.1038/nn1630
Baazaoui N, et al. Effect of CNTF Derived Peptide, P021 on Cognition and Pathology in 3xTG-AD Mouse Model of Alzheimer’s Disease.
Abstract
Studies described in this thesis deal with the preventive effects of a neurogenic/neurotropic peptidergic compound, P021, on neurogenesis and synaptic deficits, neurodegeneration, cognitive impairment, and Ab and tau pathologies in a 3xTg-AD mouse model of Alzheimer’s disease (AD).
Background: AD is a chronic progressive neurodegenerative disease. Its multifactorial nature and the heterogeneity make its treatment especially challenging. Although it is a major burden in society, at present there is no drug that can stop or slow down the progression of the disease. Currently, the only available treatments are symptomatic and for mild to severe stages. The development of a drug that can prevent AD at its early stages would be of major importance. The use of neurotrophic factors mimetic for the treatment of AD is an exciting therapeutic strategy. It focuses mainly on boosting synaptic plasticity and neurogenesis which lead to shifting the balance from neurodegeneration to regeneration of the brain as well as preventing Ab and tau pathologies. Herein, I present the preventive effects of P021 treatment in 3xTg-AD mice that is initiated very early in the disease during the period of synaptic compensation and is continued for the lifespan of the animal.
Aims: The specific aims were:
1) to study the presence of the synaptic compensation phenomenon in the brain as a self-repairing mechanism in 3xTg-AD mice;
2) to study the preventive effects of P021 on cognitive deterioration when administered during the compensation period;
3) to study the preventive effects of P021 on amyloid beta (Ab) and tau pathologies; and
4) to study the effect of P021 on synaptic deficit, neuronal degeneration and neurogenesis.
Methods: A battery of behavioral tests was conducted to assess the cognitive performance in 3xTg-AD mice with and without treatment with P021 at different disease stages. Immunohistochemical and biochemical analyses were performed to determine the levels of synaptic protein expression as well as Ab and tau pathologies at different time points that correspond to different stages of disease progression. Neurodegeneration was studied immunohistochemically with Fluorojade C staining. Neurogenesis was studied immunohistochemically with DCX (double cortin) and Ki-67 staining.
Results: The 3xTg-AD mice at the age of 12 weeks were found to be cognitively impaired and showed a decrease in multiple synaptic and neuronal markers. This decrease was compensated by the brain until ~16 weeks of age. Beyond 16 weeks the brain was found to fail to compensate for the synaptic deficit. P021 intervention, started at 14 weeks of age, prevented cognitive impairment 9 months post-treatment, as tested by the Morris Water maze task. At 15-17 months post-treatment P021 was able to rescue short-term spatial reference memory as well as episodic memory, as determined by the novel object location and the novel object recognition tasks. The treatment with P021 also prevented Ab and tau pathologies during 9-18 months post-treatment. P021 was able to rescue synaptic deficit and neurodegeneration 9-18 months post-treatment and boost neurogenesis at 9 months post-treatment. P021 treatment increased survival from 41% in 3xTg-AD-vh to 87% in 3xTg-AD-P021 mice. In the entire study I did not find any severe side effects of P021 including loss of appetite or body weight.
Conclusions: Early intervention with P021 during the period of synaptic compensation of the brain was successful in preventing cognitive impairment in 3xTg-AD mice. The P021 treatment prevented synaptic and neurogenesis deficits, neurodegeneration, and Ab and tau pathologies. These findings provide a proof of principle of the potential therapeutic effect of P021 on several major features of AD.
https://academicworks.cuny.edu/cgi/viewcontent.cgi?article=2297&context=gc_etds
Baazaoui N, et al. Prevention of dendritic and synaptic deficits and cognitive impairment with a neurotrophic compound. Alzheimers Res Ther. 2017;9:45.
Abstract
Background: The use of neurotrophic factors to treat Alzheimer’s disease (AD) is hindered by their blood–brain barrier impermeability, short half-life, and severe side effects. Peptide 021 (P021) is a neurotrophic/neurogenic tetra-peptide that was derived from the most active region of the ciliary neurotrophic factor (CNTF) by epitope mapping. Admantylated glycine was added to its C-terminal to increase its blood–brain barrier permeability and decrease its degradation by exopeptidases to make it druggable. Here, we report on the preventive effect of P021 in 3 × Tg-AD, a transgenic mouse model of AD.
Methods: P021 was administered in the diet at 3 months, i.e., 6–9 months before any overt amyloid beta (Aβ) or tau pathology, and during the period of synaptic compensation, and was continued until 21 months in 3 × Tg-AD mice. The 3 × Tg-AD mice and wild-type (WT) mice were treated identically but with a vehicle-only diet serving as controls. The effects of P021 on neurogenesis, dendritic and synaptic markers, and cognitive performance were investigated.
Results: We found that P021 treatment was able to rescue dendritic and synaptic deficits, boost neurogenesis, and reverse cognitive impairment in 3 × Tg-AD mice.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5488423/pdf/13195_2017_Article_273.pdf

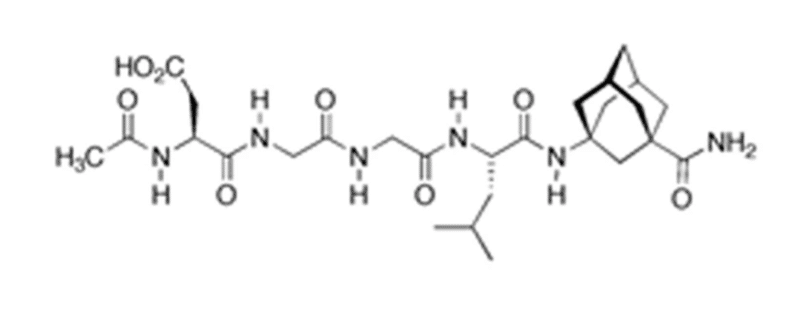
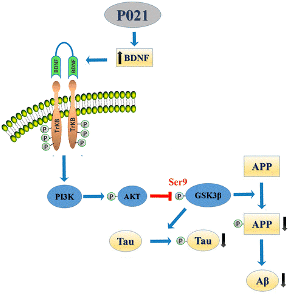 P21 (or P021) is also known as a cyclin-dependent kinase (CDK) inhibitor-1. P21 is a small tetra-peptide derived from the most active region of CNTF (ciliary neurotrophic factor), the amino acid residues 148-151. It is involved in the processes of apoptosis and cell cycle arrest and has been reported to block the activity of cyclin and CDK complexes in animal models.
P21 (or P021) is also known as a cyclin-dependent kinase (CDK) inhibitor-1. P21 is a small tetra-peptide derived from the most active region of CNTF (ciliary neurotrophic factor), the amino acid residues 148-151. It is involved in the processes of apoptosis and cell cycle arrest and has been reported to block the activity of cyclin and CDK complexes in animal models.